A Novel Inhibitor of Protease-activated Receptor 1: A Review of Chemical Structure and Mode of Action
Mehrnoosh Hashemzadeh, PhD,1,2 Joseph M. Arreguin, BS,1,2 Tyler Roberts, MS,1,2 Mohammad Reza Movahed, MD, PhD1,3
1University of Arizona Sarver Heart Center, Tucson, AZ; 2Pima Community College, Tucson, AZ; 3CareMore, Tucson, AZ
Limitations of current antiplatelet therapies have led to the discovery of new antiplatelet agents with new modes of action. Vorapaxar has been developed as a thrombin receptor antagonist. This drug works against the protease-activated receptor 1 (PAR1) and inhibits platelet aggregation mediated by PAR1. This article reviews this new class of antiplatelet therapy in detail with an acute focus on the TRACER (Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome) and TRA 2°P-TIMI 50 (Trial to Assess the Effects of Vorapaxar in Preventing Heart Attack and Stroke in Patients With Atherosclerosis-Thrombolysis In Myocardial Infarction 50) trials. Vorapaxar has proven to be beneficial when administered to stable atherosclerotic patients. However, it has been shown to increase risk of intracranial hemorrhage in patients with known, previous history of cerebrovascular incidence. Despite these limitations, TRA 2°P-TIMI 50 results showed that vorapaxar appears to have a definitive therapeutic benefit when administered alongside aspirin or when it is used as an addition to dual antiplatelet therapy for patients with stable atherosclerosis.
[Rev Cardiovasc Med. 2015;16(1):68-73 doi: 10.3909/ricm0754]
© 2015 MedReviews®, LLC
A Novel Inhibitor of Protease-activated Receptor 1: A Review of Chemical Structure and Mode of Action
Mehrnoosh Hashemzadeh, PhD,1,2 Joseph M. Arreguin, BS,1,2 Tyler Roberts, MS,1,2 Mohammad Reza Movahed, MD, PhD1,3
1University of Arizona Sarver Heart Center, Tucson, AZ; 2Pima Community College, Tucson, AZ; 3CareMore, Tucson, AZ
Limitations of current antiplatelet therapies have led to the discovery of new antiplatelet agents with new modes of action. Vorapaxar has been developed as a thrombin receptor antagonist. This drug works against the protease-activated receptor 1 (PAR1) and inhibits platelet aggregation mediated by PAR1. This article reviews this new class of antiplatelet therapy in detail with an acute focus on the TRACER (Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome) and TRA 2°P-TIMI 50 (Trial to Assess the Effects of Vorapaxar in Preventing Heart Attack and Stroke in Patients With Atherosclerosis-Thrombolysis In Myocardial Infarction 50) trials. Vorapaxar has proven to be beneficial when administered to stable atherosclerotic patients. However, it has been shown to increase risk of intracranial hemorrhage in patients with known, previous history of cerebrovascular incidence. Despite these limitations, TRA 2°P-TIMI 50 results showed that vorapaxar appears to have a definitive therapeutic benefit when administered alongside aspirin or when it is used as an addition to dual antiplatelet therapy for patients with stable atherosclerosis.
[Rev Cardiovasc Med. 2015;16(1):68-73 doi: 10.3909/ricm0754]
© 2015 MedReviews®, LLC
A Novel Inhibitor of Protease-activated Receptor 1: A Review of Chemical Structure and Mode of Action
Mehrnoosh Hashemzadeh, PhD,1,2 Joseph M. Arreguin, BS,1,2 Tyler Roberts, MS,1,2 Mohammad Reza Movahed, MD, PhD1,3
1University of Arizona Sarver Heart Center, Tucson, AZ; 2Pima Community College, Tucson, AZ; 3CareMore, Tucson, AZ
Limitations of current antiplatelet therapies have led to the discovery of new antiplatelet agents with new modes of action. Vorapaxar has been developed as a thrombin receptor antagonist. This drug works against the protease-activated receptor 1 (PAR1) and inhibits platelet aggregation mediated by PAR1. This article reviews this new class of antiplatelet therapy in detail with an acute focus on the TRACER (Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome) and TRA 2°P-TIMI 50 (Trial to Assess the Effects of Vorapaxar in Preventing Heart Attack and Stroke in Patients With Atherosclerosis-Thrombolysis In Myocardial Infarction 50) trials. Vorapaxar has proven to be beneficial when administered to stable atherosclerotic patients. However, it has been shown to increase risk of intracranial hemorrhage in patients with known, previous history of cerebrovascular incidence. Despite these limitations, TRA 2°P-TIMI 50 results showed that vorapaxar appears to have a definitive therapeutic benefit when administered alongside aspirin or when it is used as an addition to dual antiplatelet therapy for patients with stable atherosclerosis.
[Rev Cardiovasc Med. 2015;16(1):68-73 doi: 10.3909/ricm0754]
© 2015 MedReviews®, LLC
KEY WORDS
Acute coronary syndrome • Antiplatelet • Thrombin receptor antagonist • PAR1 inhibitor • Atherosclerosis • Thienopyridines • Vorapaxar
KEY WORDS
Acute coronary syndrome • Antiplatelet • Thrombin receptor antagonist • PAR1 inhibitor • Atherosclerosis • Thienopyridines • Vorapaxar
Vorapaxar works against PAR1 to inhibit platelet aggregation without affecting hemostasis.
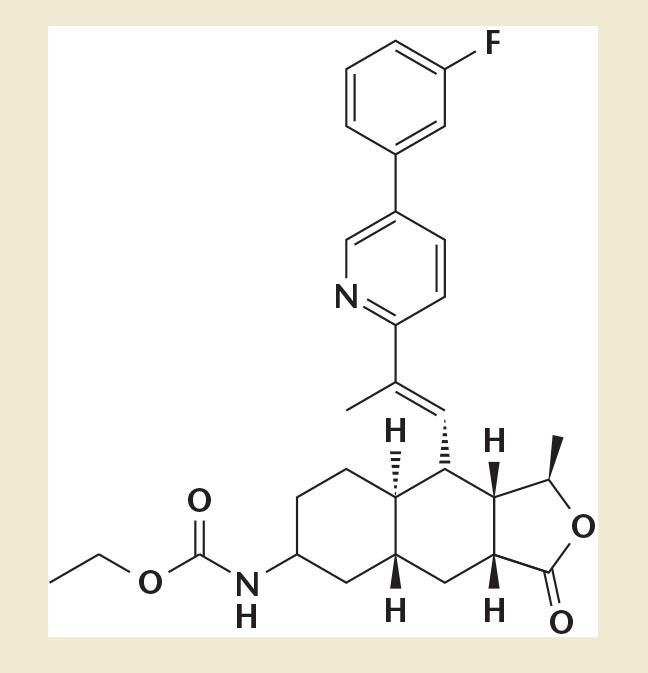
Figure 1. Chemical structure of vorapaxar.
Vorapaxar achieves over 80% inhibition of thrombin receptoractivating peptide-induced platelet aggregation within 1 week of initiation of treatment with the recommended daily dose of 2.5 mg vorapaxar sulfate.
Essentially, it is estimated that 59 patients need to be treated for 1 extra bleeding case and 100 patients need to be treated in order to reduce 1 ischemic event. Therefore, risk-to-benefit ratio should be considered in each patient before considering prescribing this new class of drug.
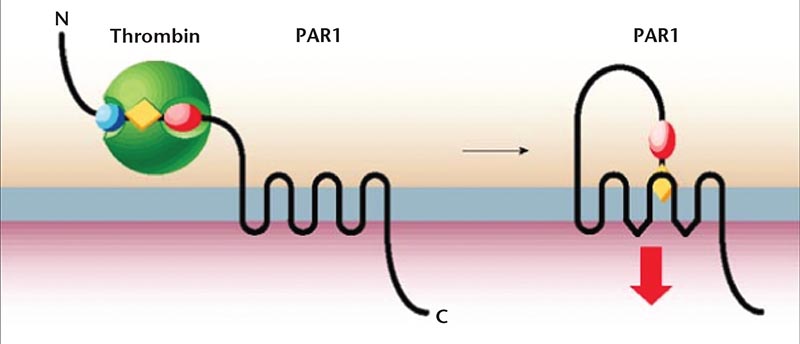
Figure 2. Cleavage of protease-activated receptor 1 (PAR1) by thrombin between arginine 41 and serine 42 exposes a new N-terminus that serves as a tethered ligand. Activation of PAR1 is followed by a rapid burst of signaling before the receptor is desensitized and, in some cases, cleared from the cell surface. Reproduced with permission from Coughlin SR.19
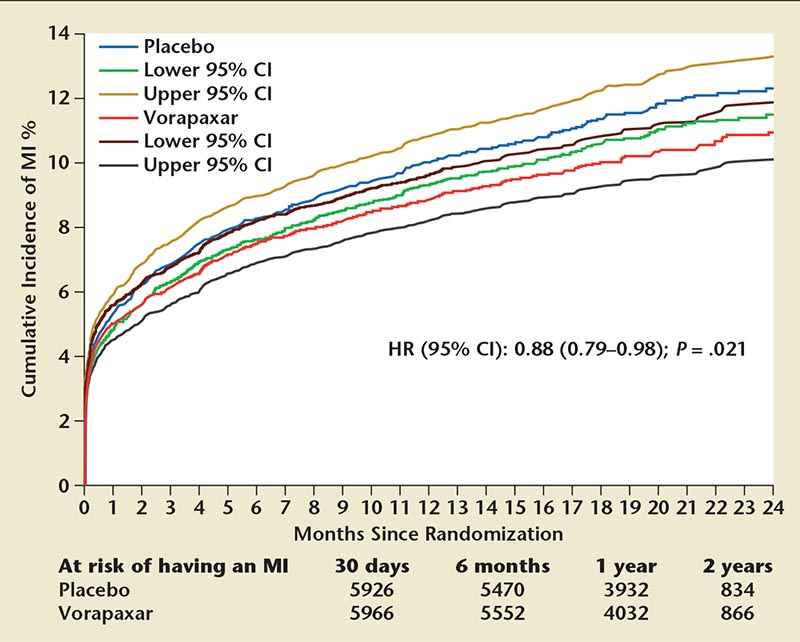
Figure 3. Incidence of any first MI with vorapaxar or placebo. Cumulative incidence with 95% CIs of a first MI of any universal MI type between vorapaxar and placebo. CI, confidence interval; HR, hazard ratio; MI, myocardial infarction. Reproduced with permission from Leonardi S et al.20
Main Points
• Antiplatelet agents are indispensable in the treatment of ischemic heart disease. However, due to their limitations, including long half-lives, a dependency on prodrug activation by the liver into the active metabolites, and persistent occurrence of stent thrombosis and acute coronary syndrome, it was important to develop another class of antiplatelet drugs.
• Protease-activated receptors (PARs) are capable of initiating platelet aggregation. They account for the majority of thrombin-induced signaling and present a strategic location for antiplatelet therapy. PAR1 is the most important receptor for thrombin response and a vital site for inhibition. Vorapaxar works against PAR1 to inhibit platelet aggregation without affecting hemostasis.
• Vorapaxar does not inhibit platelet aggregation induced by adenosine diphosphate, collagen, or a thromboxane mimetic. It is a nonprotein structure with competitive PAR1 inhibitory activity. Vorapaxar is not a prodrug, it does not need enzymatic activation, and it undergoes oxidative metabolism in the liver by the cytochrome P450 3A4 enzymes.
• Results from the Trial to Assess the Effects of Vorapaxar in Preventing Heart Attack and Stroke in Patients With Atherosclerosis-Thrombolysis In Myocardial Infarction 50 (TRA 2°P-TIMI 50) showed that vorapaxar appears to have a definitive therapeutic benefit when administered alongside aspirin or when it is used as an addition to dual antiplatelet therapy for patients with stable atherosclerosis with some increasing risk for bleeding.
Main Points
• Antiplatelet agents are indispensable in the treatment of ischemic heart disease. However, due to their limitations, including long half-lives, a dependency on prodrug activation by the liver into the active metabolites, and persistent occurrence of stent thrombosis and acute coronary syndrome, it was important to develop another class of antiplatelet drugs.
• Protease-activated receptors (PARs) are capable of initiating platelet aggregation. They account for the majority of thrombin-induced signaling and present a strategic location for antiplatelet therapy. PAR1 is the most important receptor for thrombin response and a vital site for inhibition. Vorapaxar works against PAR1 to inhibit platelet aggregation without affecting hemostasis.
• Vorapaxar does not inhibit platelet aggregation induced by adenosine diphosphate, collagen, or a thromboxane mimetic. It is a nonprotein structure with competitive PAR1 inhibitory activity. Vorapaxar is not a prodrug, it does not need enzymatic activation, and it undergoes oxidative metabolism in the liver by the cytochrome P450 3A4 enzymes.
• Results from the Trial to Assess the Effects of Vorapaxar in Preventing Heart Attack and Stroke in Patients With Atherosclerosis-Thrombolysis In Myocardial Infarction 50 (TRA 2°P-TIMI 50) showed that vorapaxar appears to have a definitive therapeutic benefit when administered alongside aspirin or when it is used as an addition to dual antiplatelet therapy for patients with stable atherosclerosis with some increasing risk for bleeding.
Antiplatelet agents are indispensable in the treatment of ischemic heart disease. Aspirin was used as a lone anti-platelet therapy until the discovery of a new group of antiplatelet drugs called thienopyridines. When administered together, aspirin and thienopyridines (eg, clopidogrel) have been found to improve the outcome of patients presenting with acute coronary syndrome (ACS). They are essential in the dual antiplatelet therapy for reducing the risk of stent thrombosis.1 Clopidogrel acts as a P2Y12 inhibitor, blocking adenosine diphosphate (ADP) receptors, thus preventing platelet aggregation.2 Due to the limitations of these drugs, which include long half-lives, a dependency on pro-drug activation by the liver into the active metabolites, and persistent occurrence of stent thrombosis and ACS, it was important to develop another class of antiplatelet drugs.3 Ticagrelor, an oral antiplatelet medication that is a reversible inhibitor of P2Y12, has a shorter half-life and does not require liver activation. Its ability to initiate action in less than 1 hour further contrasts its predecessors. Ticagrelor was approved by the US Food and Drug Administration (FDA) in 2011.4
None of these drugs, however, inhibit the most crucial platelet receptor proteins. Protease-activated receptors (PARs), alone, are capable of initiating platelet aggregation. They account for the majority of thrombin-induced signaling and present a strategic location for antiplatelet therapy.5 Consequently, in order to have an additional antiplatelet agent with this new mode of action, vorapaxar—a thrombin receptor antagonist—was developed. PAR1 is the most important receptor for thrombin response. Vorapaxar works against PAR1 to inhibit platelet aggregation without affecting hemostasis. Vorapaxar is absorbed orally and metabolized and eliminated primarily through gastrointestinal routes.6
In 2007, the Thrombolysis in Myocardial Infarction (TIMI) Study Group at Brigham and Women's Hospital (Boston, MA) and Duke Clinical Research Institute (Durham, NC) each launched an experimental trial in an attempt to validate vorapaxar and its efficacy. Their two trials, Trial to Assess the Effects of Vorapaxar in Preventing Heart Attack and Stroke in Patients With Atherosclerosis-Thrombolysis In Myocardial Infarction 50 (TRA 2°P-TIMI 50) and Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome (TRACER), respectively, have been enrolled in approximately 2000 centers and over 30 countries combined. In patients presenting with ACS without ST-segment elevation, the randomized, multicenter, double-blinded, placebo-controlled TRACER study was conducted to evaluate the effects of this thrombin receptor antagonist when administered in addition to dual antiplatelet treatment.7 After recommendations made by the Data and Safety Monitoring Board, the trial was ended early due to higher risk for intracranial bleeding in patients with previous history of stroke.
The concomitant multicenter, randomized, double-blinded, placebo-controlled TRA 2°P-TIMI 50 study was conducted on stable patients with known history of ath-erothrombosis.8 This trial showed that vorapaxar, when added to standard antiplatelet therapy, reduced mortality in patients with a history of myocardial infarction (MI) or stroke with the risk of increased moderate or severe bleeding over dual antiplatelet therapy. Patients who had previous stroke history discontinued the trial after 2 years because of a significant increase in intracerebral hemorrhage. Vorapaxar received its FDA approval in the United States on May 5, 2014, as a once-daily tablet for the reduction of thrombotic cardiovascular events in patients with a history of MI or peripheral vascular disease.9
Chemical Structure and Mechanism of Action
The classes of antiplatelet drugs that have proven efficacious include aspirin, thienopyridines (eg, ticlopidine, clopidogrel, prasugrel), nonthieno-pyridines (eg, ticagrelor), and glyco-protein Ilb/IIIa receptor antagonists (eg, abciximab, eptifibatide, tirofiban). Novel antiplatelet agents that conquer some of the potential limitations of these agents are in development and are currently being marketed after completion of two multicenter clinical trials. Vorapaxar is an oral agent with a high selectivity for PAR1 inhibition and does not interfere with fibrinogen cleavage.10 Vorapaxar sulfate has a chemical name of ethyl [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-{(1E)-2-[5-(3-fluorophenyl)pyridin-2-yl]ethen-1-yl}-1-methyl-3-oxododecahydronaphtho[2,3-c]furan-6-yl]carbamate sulfate and a molecular formula of C29H33FN2O4-H2SO4. Vorapaxar is available for oral use as a tablet containing 2.08 mg of vorapaxar, which is equivalent to 2.5 mg of vorapaxar sulfate. Partial conversion of vorapaxar sulfate to free vorapaxar may occur during the manufacturing and storage periods.
Vorapaxar does not inhibit platelet aggregation induced by ADP, collagen, or a thromboxane mimetic. It is a nonprotein structure with competitive PAR1 inhibitory activity (Figure 1). It is a synthetic tricyclic 3-phenylpyridine analog of himbacine, an alkaloid isolated from the bark of Australian magnolias.11 Vorapaxar is not a pro-drug, it does not need enzymatic activation, and it undergoes oxidative metabolism in the liver by the cytochrome P450 3A4 (CYP3A4) enzymes. The liver excretes 90% as inactive metabolites. Therefore, it is recommended to avoid any strong CYP3A4 inhibitors (eg, ketoconazole, itraconazole) while taking vorapaxar. CYP3A4 inducers (eg, carbamazepine, rifampin, St. John's Wort, phenytoin) should also be avoided. Its major active circulating metabolite is M20 (monohydroxy metabolite), and the predominant metabolite identified in excretion is Ml9 (amine metabolite).
Vorapaxar achieves over 80% inhibition of thrombin receptor-activating peptide-induced platelet aggregation within 1 week of initiation of treatment with the recommended daily dose of 2.5 mg vorapaxar sulfate. The duration of platelet inhibition is dose and concentration dependent. Studies have shown that the inhibition of platelet aggregation at a level of 50% can be expected up to 4 weeks after discontinuation of daily dosing. This is consistent with the terminal elimination half-life of the drug (Figure 1).
The most potent platelet agonist naturally produced is thrombin. Thrombin-mediated platelet activation plays a critical role in the pathophysiology of thrombosis.12 Consequently, thrombin activation has been a focal point of recent research. It has been revealed that thrombin activates a number of cell types via proteolytic activation of specific G protein-coupled cell surface receptors known as PARs.13 There are four known types of PARs, among which the prototypical PAR1 receptor, also known as a thrombin receptor, is widely distributed in human platelets, endothelial cells, and smooth muscle cells. PAR1 is activated when thrombin binds and cleaves its amino-terminal exodomain to unmask a new receptor amino terminus. This new amino terminus is then available as a peptide-tethered ligand that binds to the receptor intramolecularly in order to enable signaling across the membrane.
The thrombin receptor PAR1 belongs to the family of seven transmembrane G protein-coupled receptors. The mechanism of PAR1 activation by thrombin (discovered by Vu and colleagues14 in 1991) is unique. Current evidence suggests that thrombin activates human platelets by cleaving and activating its receptor hirudin-like site, which acts as a thrombin-binding site. It binds to PAR1 through its exo-anion binding site and causes the cleavage of the extracellular domain at arginine-41/serine-42 and consequently reveals a new protonated amino group at the NH2-terminus of tethered ligand: S42FLLRN (ser-ine -phenylalanine -leucine-leucine -arginine-asparagine). This tethered ligand then binds intramolecularly (Figure 2).14
Clinical Trials
Two phase II clinical trials were completed to assess (1) the risk of bleeding and (2) overall efficacy. The first, an early phase II trial, was performed to assess the outcome of patients who underwent nonurgent percutaneous coronary intervention (PCI), as well as to evaluate the bleeding risk, toler-ability, and efficacy of different doses (loading and maintenance) of vorapaxar (and aspirin, 94%). Clopidogrel was used concomitantly in 67% of patients. The trial showed that the drug was well tolerated without an increase in TIMI bleeding when added to a dual anti-platelet regimen. The primary end-point in the trial was TIMI-defined major or minor bleeding. TIMI major or minor bleeding occurred at a similar rate as in the placebo patients (3% vs 3%; -0.5% difference for patients with vorapaxar); however, with a small patient pool (n = 673) the results were not statistically significant.15 The second, a Japanese phase II trial in patients presenting with non-ST segment elevation ACS, tested vorapaxar against a placebo in addition to aspirin, ticlopidine, and heparin. The primary safety endpoint was TIMI major or minor bleeding. The trial found an insignificant increase in TIMI major or minor bleeding when compared with placebo (14% vs 10%). Periprocedural MI had a significantly lower rate in the vorapaxar group (16.9% vs 42.9%), although, like the previous phase II trial, the patient pool was small (n = 92) and required further study.16
Subsequently, two phase III trials were conducted to evaluate the safety and efficacy of vorapaxar in different sets of patients. The TRACER trial included patients with symptoms of ACS within 24 hours of hospitalization and at least one of the following: age ≥ 55 years; history of MI, PCI, coronary bypass grafting (CABG), diabetes, or peripheral arterial disease.7 In this trial, vorapaxar was tested against a placebo, in addition to both aspirin and clopidogrel.
The second trial, TRA 2°P-TIMI 50, included patients with either spontaneous MI or incident of ischemic stroke within 2 weeks to 12 months prior to enrollment.8 In this trial, vorapaxar was tested against a placebo with 93% of patients taking aspirin and 62% taking aspirin along with a thienopyridine. The TRACER trial used a revised primary endpoint that included cardiovascular death, stroke, MI, urgent revascularization, and recurrent ischemia. The secondary endpoints for TRACER included cardiovascular death, stroke, or MI. These were the primary endpoints in TRA 2°P-TIMI 50, whereas cardiovascular death, stroke, MI, urgent revascularization, and recurrent ischemia were secondary end-points (Figure 3).
Clinical Trials in Patients Treated Conservatively
Efficacy
In TRACER (N = 12,944), the primary efficacy endpoint occurred in 18.5% of patients taking vorapaxar. This compares with 19.9% of patients taking a placebo (hazard ratio [HR] 0.92, 95% confidence interval, 0.85-1.01; P = .O7). The key secondary efficacy endpoint occurred in 14.7% of vorapaxar-treated patients and 16.4% of placebo-treated patients (HR 0.89; P = .02). Stroke occurred at approximately the same rate (1.9% vs 2.1%). However, hemorrhagic stroke occurred in 0.3% of vorapaxar patients and 0.1% of placebo patients (HR 2.73; P = .02)7 This led to premature discontinuation of this trial.
In TRA 2°P-TIMI 50 (N = 26,449), the primary efficacy end-point occurred in 11.2% of vorapaxar patients and 12.4% of placebo patients (HR 0.88; P = .001). The secondary efficacy endpoint occurred in 9.3% and 10.5% of patients, respectively (HR 0.87; P < .001). Stroke occurred at a rate of 2.8% in both arms of the study (HR 0.97; P = .73).
Adverse Events
Bleeding in TRACER was measured against both Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries (GUSTO) and TIMI criteria. GUSTO moderate or severe bleeding occurred in 7.2% of vorapaxar patients and 5.2% of placebo patients (HR 1.35; P < .001). Severe bleeding occurred at a rate of 2.9% and 1.6%, respectively. Under the TIMI criteria, clinically significant bleeding occurred in 20.2% of vorapaxar and 14.6% of placebo patients (HR 1.43; P < .001). Major bleeding comprised 4.0% and 2.5% of these totals. Additionally, intra-cranial hemorrhage occurred in 1.1% of vorapaxar- and 0.2% of placebo-treated patients (HR 3.39; P < .001). Fatal bleeding and bleeding that contributed to death occurred in 12 vorapaxar patients and 4 placebo patients.
Bleeding was also measured with GUSTO and TIMI criteria in TRA 2°P-TIMI 50, but without a breakdown of moderate to severe bleeding in GUSTO or significant and major bleeding in TIMI. GUSTO moderate or severe bleeding occurred in 4.2% of vorapaxar patients and 2.5% of placebo patients (HR 1.66; P < .001). Essentially, it is estimated that 59 patients need to be treated for 1 extra bleeding case and 100 patients need to be treated in order to reduce 1 ischemic event. Therefore, risk-to-benefit ratio should be considered in each patient before considering prescribing this new class of drug. TIMI clinically significant bleeding occurred in 15.8% of vorapaxar patients and 11.1% of placebo patients (HR 1.46; P < .001). Fatal bleeding occurred in 29 vorapaxar patients and 20 placebo patients.8
Although both TRACER and TRA 2°P-TIMI 50 compared favorably with placebo with regard to efficacy, they both showed increased risk of bleeding. In particular, patients in TRACER had a much higher occurrence of intracranial and fatal bleeding. Intracranial and fatal bleeding occurred at a higher rate in TRA 2°P-TIMI 50, but paralleled a similarly higher rate in placebo trials (0.6% vs 0.4%; HR 1.54; P = .076 and 0.2% vs 0.1%; HR 1.56; P = .3, respectively).17 The major differences in the results of TRACER and TRA 2°P-TIMI 50 are due to the differences in patient selection. TRACER included patients with ACS, including patients who had undergone PCI and CABG (23% and 12%, respectively). Additionally, all patients in TRACER experienced an ischemic event within 24 hours of hospital presentation. Conversely, patients in TRA 2°P-TIMI 50 were stable with a history of MI or ischemic stroke within 2 weeks to 12 months. The decrease in primary and secondary endpoints in TRACER was insignificant. Furthermore, the patients had increased bleeding. It should also be noted that patients with a lower body weight (< 60 kg) experienced GUSTO moderate to severe bleeding at a higher rate. Additionally, patients who had previously taken thienopyridines had a higher risk of bleeding than those who began a thienopyridine at randomization.
In January 2011, the Data and Safety Monitoring Board discovered a high rate of intracranial bleeding in patients with stroke history. As a result, such patients were removed from the trial and separate analyses were carried out only for patients with a history of peripheral arterial disease and MI. Within this subgroup, there was a positive net clinical outcome for stable atherosclerotic patients. Especially of note, there was a 20% decrease in risk for patients with a history of MI.14 The findings of the trial also included increased bleeding for patients < 60 kg and age > 75 years.7
Conclusions
Vorapaxar, definitively, has shown benefit when administered to stable atherosclerotic patients on dual or single antiplatelet therapy. In patients presenting with ACS, there were not sufficient data to draw conclusions, as the TRACER trial was discontinued early with higher intracranial bleeding in patients with history of stroke. Additionally, for patients with stable atherosclerosis, age and weight should be considered to decrease the risk of bleeding. These bleeding risks and the risk of intracranial bleeding also significantly increase for patients with a history of stroke. Patients with a history of cerebrovascular incidence should avoid the use of vorapaxar. Despite these limitations, based on the results of TRA 2°P-TIMI 50, vorapaxar appears qualified to be used in association with aspirin or in addition to dual antiplatelet therapy for patients with stable atherosclerosis. Future trials should be conducted to compare vorapaxar with clopidogrel or other thienopyridines. This would provide contrast to the TRA 2°P-TIMI 50 trial, which tested vorapaxar in addition to dual antiplatelet therapy. This comparison would potentially reduce the risk of bleeding in the vorapaxar arm.
Another PAR1 antagonist, atopaxar, has been studied in the phase II Japanese Lessons from Antagonizing the Cellular Effect of Thrombin (J-LANCELOT) trials. The results indicated a reduction of major cardiovascular adverse events in ACS patients. However, bleeding events were found to increase in atopaxar-treated patients compared with those treated with placebo. Furthermore, liver enzymes were elevated and the relative, corrected QT interval was prolonged in atopaxar-treated patients. Consequently, development of atopaxar was discontinued.
Vorapaxar has not yet been reviewed for antiplatelet monotherapy; it was administered only as an addition to aspirin and/or clopidogrel. It would be interesting to per form studies juxtaposing the individual efficacy of vorapaxar with that of aspirin and/or clopidogrel. Future studies on thrombin inhibi tors could also involve studying the effects of these drugs on matrix metalloproteinases, which have been previously shown to activate PAR1 receptors in human cells.18 ![]()
The authors report no real or apparent conflicts of interest.
References
- Khoynezhad A, Movahed MR, Hashemzadeh M, et al. Perioperative management of patients on adenosine diphosphate inhibitors in the era of drug-eluting stents: review of the literature and clinical implications. Curr Med Chem. 2009;16:591-598.
- Hashemzadeh M, Goldsberry S, Furukawa M, et al. ADP receptor-blocker thienopyridines: chemical structures, mode of action and clinical use. A review. J Invasive Cardiol. 2009;21:406-412.
- Springthorpe B, Bailey A, Barton P, et al. From ATP to AZD6140: the discovery of an orally active reversible P2Y12 receptor antagonist for the prevention of thrombosis. Bioorg Med Chem Lett. 2007;17:6013-6018.
- Radhakrishna R, Movahed MR, Hashemzadeh M. Novel antiplatelet agent ticagrelor in the management of acute coronary syndrome. J Interv Cardiol. 2011;24:199-207.
- Rivera J, Lozano M, Navarro-Nuñez L, Vicente V. Platelet receptors and signaling in the dynamics of thrombus formation. Haematologica. 2009;94: 700-711.
- Iannopollo G, Camporotondo R, De Ferrari G, Leonardi S. Efficacy versus safety: the dilemma of using novel platelet inhibitors for the treatment of patients with ischemic stroke and coronary artery disease. Ther Clin Risk Manag. 2014;10:321-329.
- Tricoci P, Huang Z, Held C, et al; TRACER Investigators. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med. 2012;366:20-33.
- Morrow D, Braunwald E, Bonaca MP, et al; TRA 2P-TIMI 50 Steering Committee and Investigators. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366:1404-1413.
- US Food and Drug Administration. FDA approves Zontivity to reduce the risk of heart attacks and stroke in high-risk patients [news release]. May 8, 2014. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm396585.htm. Accessed January 15, 2015.
- Doller D, Chackalamannil S, Czarniecki M, et al. Design, synthesis, and structure-activity relationship studies of himbacine-derived muscarinic receptor antagonists. Bioorg Med Chem Lett. 1999;9: 901-906.
- Chackalamannil S, Wang Y, Greenlee WJ, et al. Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity. J Med Chem. 2008;51:3061-3064.
- Sambrano GR, Weiss EJ, Zheng YW, et al. Role of thrombin signaling in platelets in hemostasis and thrombosis. Nature. 2001;413:74-78.
- Coughlin SR. How the protease thrombin talks to cells. Proc Natl Acad Sci U S A. 1999;96: 11023-11027.
- Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057-1068.
- Becker RC, Moliterno DJ, Jennings LK, et al; TRA-PCI Investigators. Safety and tolerability of SCH 530348 in patients undergoing non-urgent percutaneous coronary intervention: a randomized, double-blind, placebo controlled phase II study. Lancet. 2009;373: 919-928.
- Goto S, Yamaguchi T, Ikeda Y, et al. Safety and exploratory efficacy of the novel thrombin receptor (PAR-1) antagonist SCH530348 for non-ST-segment elevation acute coronary syndrome. J Atheroscler Thromb. 2010;17:156-164.
- Goto S, Tomita A. New antithrombotics for secondary prevention of acute coronary syndrome. Clin Cardiol. 2014;37:178-187.
- Trivedi V, Boire A, Tchernychev B, et al. Platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell. 2009;137:332-343.
- Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258-264.
- Leonardi S, Tricoci P, White HD, et al. Effect of vorapaxar on myocardial infarction in the thrombin receptor antagonist for clinical event reduction in acute coronary syndrome (TRA·CER) trial. Eur Heart J. 2013;34:1723-1731.