Improvement of Melt Strength and Crystallization Rate of Polylactic Acid and its Blends
…with medium-chain-length polyhydroxyalkanoate through reactive modification
Previous Article Next Article
By Manoj Nerkar1, Juliana Ramsay1, Marianna Kontopoulou1, and Bruce Ramsay2
1Chemical Engineering, Queen’s University, Kingston, Ontario, Canada
2PolyFerm Canada, Kingston, Ontario, Canada
Improvement of Melt Strength and Crystallization Rate of Polylactic Acid and its Blends
…with medium-chain-length polyhydroxyalkanoate through reactive modification
Previous Article Next Article
By Manoj Nerkar1, Juliana Ramsay1, Marianna Kontopoulou1, and Bruce Ramsay2
1Chemical Engineering, Queen’s University, Kingston, Ontario, Canada
2PolyFerm Canada, Kingston, Ontario, Canada
Improvement of Melt Strength and Crystallization Rate of Polylactic Acid and its Blends
…with medium-chain-length polyhydroxyalkanoate through reactive modification
Previous Article Next Article
By Manoj Nerkar1, Juliana Ramsay1, Marianna Kontopoulou1, and Bruce Ramsay2
1Chemical Engineering, Queen’s University, Kingston, Ontario, Canada
2PolyFerm Canada, Kingston, Ontario, Canada
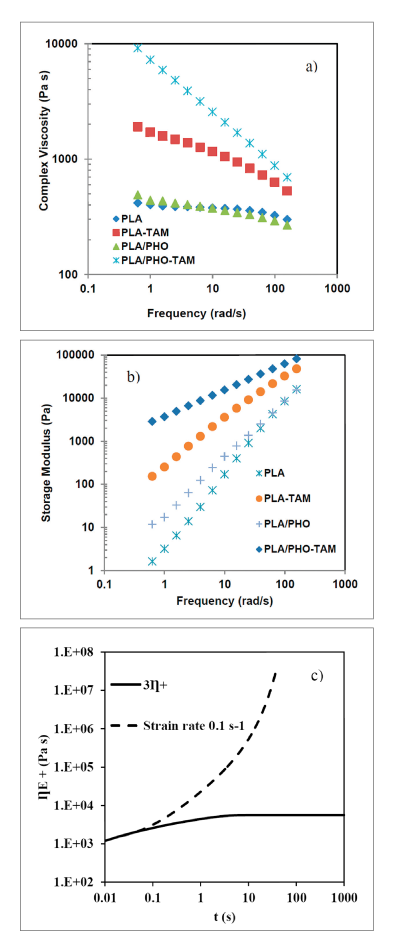
Figure 1: a) complex viscosity and b) storage modulus as a function of frequency at 180°C; c) extensional stress growth at 180°C
Figure 1: a) complex viscosity and b) storage modulus as a function of frequency at 180°C; c) extensional stress growth at 180°C
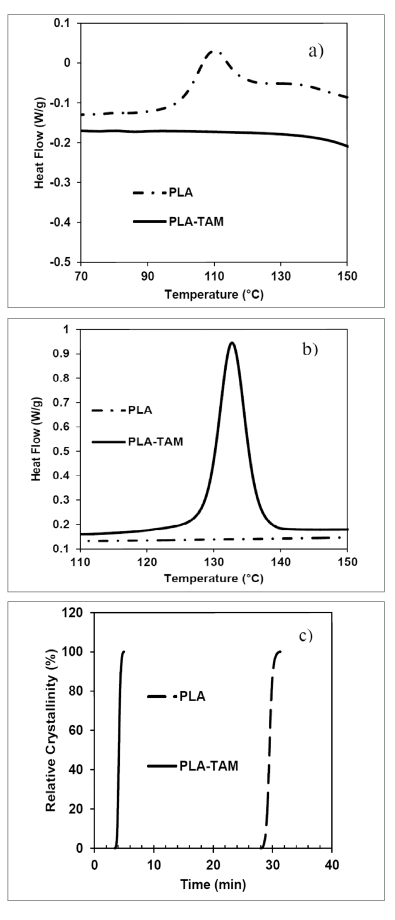
Figure 2: DSC a) heating scan at rate of 5°C/min., b) cooling scan at the rate of 5°C/min., and c) relative crystallinity at the cooling rate of 20°C/min.
Figure 2: DSC a) heating scan at rate of 5°C/min., b) cooling scan at the rate of 5°C/min., and c) relative crystallinity at the cooling rate of 20°C/min.
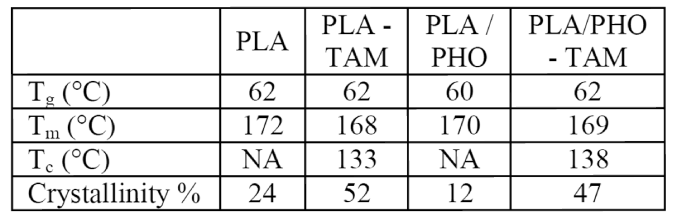
Table 1: Thermal Characterization of PLA and PLA/PHO Blends
Table 1: Thermal Characterization of PLA and PLA/PHO Blends
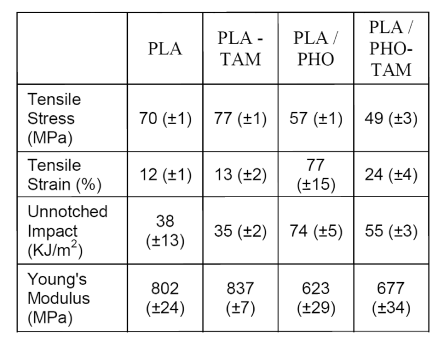
Table 2: Mechanical Properties of PLA and PLA/PHO Blends
Table 2: Mechanical Properties of PLA and PLA/PHO Blends

Figure 3: Scanning electron microscopic images of a) PLA/PHO (90/10) blend and b) co-agent-modified PLA/PHO (90/10) blend
Figure 3: Scanning electron microscopic images of a) PLA/PHO (90/10) blend and b) co-agent-modified PLA/PHO (90/10) blend
Introduction
Poly(lactic acid) (PLA) is widely used as an alternative to petroleum-based polymers, because it is a biodegradable polymer derived from renewable sources and can be processed using conventional polymer-processing techniques. Key challenges that restrict PLA from gaining more commercial acceptance include its brittle nature and low crystallization rates.
Blending with ductile polymers and addition of plasticizers and nucleating agents are commonly used to overcome these limitations. Zaman et al. used a thermoplastic polyester elastomer to impact-modify PLA.1 Linear low-density polyethylene, high-density polyethylene, polyethylene oxide, and polycaprolactone have been blended with PLA to reduce its brittleness.1
Nucleating agents are commonly used to overcome slow crystallization rates and to increase the overall crystallinity, while maintaining small crystal sizes to ensure good mechanical properties. Some common nucleating agents include talc, mica, calcium carbonate, chalk, and boron nitride. Nofar et al.2 evaluated micro-sized talc, nanosilica, and nanoclays to nucleate PLA. Talc resulted in faster nucleation and increased crystallinity. On the contrary, nanoclay and nanosilica delayed nucleation and hindered crystal growth, resulting in lower crystallinity compared to a PLA-talc system. Biobased nucleating agents such as cellulose nanocrystals have been used to increase the crystallization rate of PLA without compromising its environmentally friendly and biocompatible nature.3
Polyhydroxyalkanoates (PHA) are linear polyesters derived from renewable resources. They are typically produced by microorganisms using a fermentation process. They can be categorized into three groups: short-chain-length (SCL PHA), medium-chain-length (MCL PHA), and hybrids of MCL and SCL PHA. MCL PHAs have 3-11 carbons in their side chain, whereas SCL PHAs have only 0-2 carbons.1 MCL PHAs have low crystallinity and exhibit elastomeric properties. They are promising biopolymers as impact modifiers for PLA.
In our previous work,1 we demonstrated that addition of polyhydroxyoctanoate (PHO), which is an MCL PHA, to PLA resulted in modest improvements in the impact properties of PLA. However, the morphology of these blends was very coarse because of the very low melt viscosity of PHO, suggesting the need for chain extension for these materials to improve their rheological properties.
Reactive extrusion has been used to improve the properties of biopolymer blends. Praphulla et al.4 followed a reactive extrusion approach, using dicumylperoxide (DCP) and a multifunctional co-agent trimethylol propanetrimethacrylate (TMPTA) to produce polyhydroxybutyrate-co-valerate (PHBV) and polybutylene succinate (PBS) blends. They suggested that this reaction enhanced the compatibility between the two polymers. You et al.5 used pentaerythritol triacrylate (PETA) and bis (1-methyl-1-phenylethyl) peroxide for the reactive extrusion of PLA. Modified PLA showed increased crystallization rate with substantial decrease in the crystallization half time.
In the present work, reactive extrusion of PLA and PLA/PHO blend was conducted using a multi-functional co-agent triallyl trimesate (TAM) and DCP. The thermal, rheological, mechanical, and morphological properties of the reactively modified PLA and PLA/PHO blends are discussed.
Experimental
PLA (3251D, MFR: 35g/10 min (190°C, 2.16 kg)) was obtained from NatureWorks LLC. PHO containing ~98 mol% of 3-hydroxyoctanoate and ~2 mol% of 3-hydroxyhexanoate was supplied by Polyferm Canada. TAM and DCP were obtained from Monomer-Polymer and Dajac Labs, and Sigma Aldrich, respectively, and used as received. Based on DSC (differential scanning calorimeter) characterization, performed on a Q100 DSC from TA instruments, PHO has a melting peak at 63°C, crystallinity of 15%,1 and a Tg at -38°C (first heating scan); there is no melting transition during the second scan. The crystallinity of PLA and the PLA/PHO blends was determined using Equation 1:
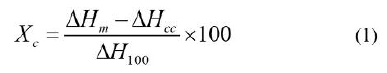
where ΔHm is the apparent fusion enthalpy, ΔHcc is the exothermic enthalpy that is absorbed by crystals formed during the heating scan, and ΔH100 is the theoretical fusion enthalpy of a 100% crystalline polymer, which is 93 J/g1 for PLA and 98.3 J/g, calculated theoretically for the blend.
PLA and blends of PLA containing 10 wt% PHO were reacted with 0.3 wt% DCP and 1 wt% TAM using a DSM micro-compounder at 180°C, to produce PLA-TAM and PLA/PHO-TAM derivatives. PLA and PHO were dried in a vacuum oven at 100°C and at room temperature, respectively, to remove moisture. The linear viscoelastic properties were measured in the oscillatory mode using a stress-controlled rheometer (Visco Tech, Reologica). Frequency sweeps were conducted at 180°C using 20-mm parallel plates. Samples were further characterized in uniaxial extension using an SER-2 universal testing platform from Xpansion Instruments hosted on an MCR-301 Anton Paar rheometer. Measurements were conducted at 180°C at extension rates ranging from 0.10 to 10 s-1.
Compression-molded specimens were used for mechanical characterization. Tensile testing was conducted on an Instron 3369 in accordance with ASTM D638. Izod impact strength measurements were carried out on unnotched specimens according to ISO 180. Blend morphologies were observed using a JEOL JSM-840 scanning electron microscope. Samples were first hot-pressed at 200°C for 3 min., then immersed in liquid nitrogen for 3 min. before brittle fracture.
Results and Discussion
Reactive extrusion of PLA in the presence of TAM and DCP resulted in a substantial increase in the viscosity and elasticity of PLA and PLA/PHO blends (Figures 1a) and 1b)), suggesting that the free radicals generated by DCP interact with the multifunctional co-agent TAM to form a branched structure. The presence of extensive branching is also evident from the presence of shear thinning (Figure 1a)) and the increase in the extensional viscosity (Figure 1c)).
Whereas PLA is known to have low melt strength and does not show strain hardening, co-agent modification of PLA led to substantial strain hardening, suggesting improved melt strength. Furthermore, as shown in Figure 1, the increase in viscosity is even higher in PLA/PHO blend than pristine PLA, despite the very low viscosity of PHO. This suggests the presence of substantial interactions between the two phases upon reactive modification. There is also the possibility that the DCP and co-agent react preferentially with the PHO rather than PLA.
The crystallization behavior of co-agent-modified PLA was studied using DSC. Detailed DSC data is presented in Table 1. Typically pristine PLA has a cold crystallization peak at ~110°C (Figure 2a)) and does not have a crystallization peak during the cooling cycle. On the contrary, co-agent-modified PLA showed a clear crystallization peak at 133°C (Figure 2b)). Similar observations were made for the PLA/PHO blend (Table 1). This points to a nucleating effect, even though no nucleating agent was added. Recently, a similar nucleating effect has been reported in co-agent-modified polypropylene, and has been attributed to the formation of a separate phase of co-agent-rich particles that forms upon reactive modification.6
The crystallinity of PLA was enhanced by 114%, and the increase in crystallinity of PLA/PHO blend was 274%, indicating a strong nucleation effect. Additionally, co-agent modification led to the disappearance of the cold crystallization peak. No significant change was observed in the glass transition temperatures, while the melting temperature of modified PLA was reduced by 4°C.
The relative crystallinity as a function of time, recorded during non-isothermal crystallization experiments, is shown in Figure 2c). While cooling from the melt state at a rate of 20°C/min., modified PLA started to crystallize significantly earlier as compared to pristine PLA. However, the crystallization half-time of modified PLA was higher (0.6 min.) than that of pristine PLA (0.38 min.).
The effect of co-agent on the mechanical properties of PLA and PLA/PHO blends is shown in Table 2. Co-agent modification did not affect the properties of PLA significantly, except for the tensile stress, which increased by 10%. The PLA/PHO blend containing 10 wt% PHO had enhanced tensile strain and unnotched impact strength compared to PLA, confirming that PHO acts as an impact modifier. The tensile strain and unnotched impact strength of the co-agent-modified PLA/PHO blend were not as high as the unmodified blend’s; however, they were better than the neat PLA’s.
Figure 3 shows scanning electron microscope (SEM) images of PLA/PHO blends. Co-agent modification reduced the size of the dispersed PHO domains, leading to significantly improved morphology of the blend.
The gel content of co-agent-modified PLA and PLA/PHO blend was 1 and 20%, respectively, revealing the presence of cross-linked chains in the modified blend. These gels may compromise the ductility of the blend, thus explaining the drop in ductility and impact strength compared to the unmodified blend.
Conclusions
PLA and its blend with MCL PHA were reacted in-situ with a multifunctional co-agent, TAM, in the presence of DCP. Co-agent-modified PLA and its blends with PHO had significantly higher melt viscosity than the non-reacted polymers. Significant strain hardening was achieved in modified PLA. Appearance of a crystallization peak in co-agent-modified PLA indicated a crystallization nucleation effect. Modified PLA maintained the mechanical properties of neat PLA. Addition of 10% of PHO in PLA improved impact strength and percent elongation of PLA. The morphology of the PLA/PHO blend was improved with co-agent modification.
Acknowledgments
Funding from the Natural Sciences and Engineering Council (NSERC) of Canada and Xerox Research Center of Canada is gratefully acknowledged.
References
- Nerkar M., Ramsay J., Ramsay B., Kontopoulou M. Polymer Processing Society, Germany, 2013.
- Nofar M., Tabatabaei A., Park C.B. Polymer 2013; 54(9):2382-2391.
- Pei A., Zhou Q., Berglund L.A. Composites Sci Technol 2010; 70(5):815-821.
- Praphulla P., Mohanty A., Misra M. Polymer Processing Society, Canada, 2012.
- You J., Lou L., Yu W., Zhou C. J Appl Polym Sci 2013; 129(4):1959-1970.
- Zhang Y., Tiwary P., Parent J.S., Kontopoulou M., Park C.B. Polymer 2013; 54(18):4814-4819.