Maintaining High Standards for Medical Packaging
The key is to keep up these standards throughout the quality testing process
Previous Article Next Article
By Elayne Schneebacher
Instron, Norwood, Massachusetts, USA
Maintaining High Standards for Medical Packaging
The key is to keep up these standards throughout the quality testing process
Previous Article Next Article
By Elayne Schneebacher
Instron, Norwood, Massachusetts, USA
Maintaining High Standards for Medical Packaging
The key is to keep up these standards throughout the quality testing process
Previous Article Next Article
By Elayne Schneebacher
Instron, Norwood, Massachusetts, USA
Usability peel test for medical packaging.

Ball burst test on packaging film.

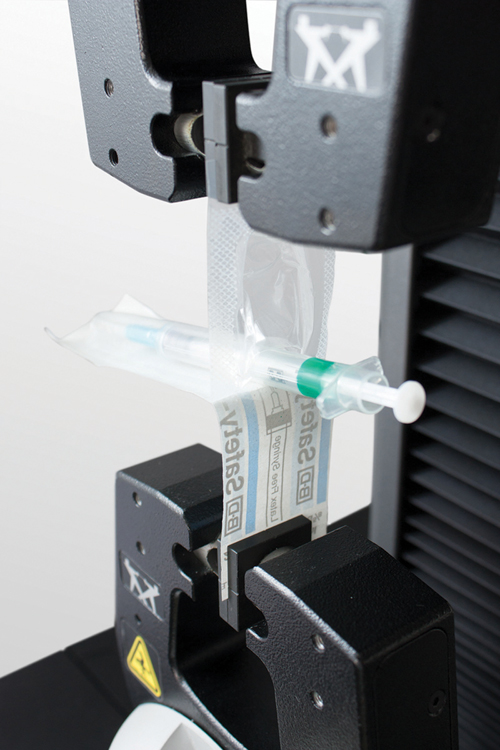
Testing pull-out force on a medical intravenous therapy pouch.

Aspiration and ejection testing to ISO 7885-1.
The integrity of medical packaging is often evaluated with equal attention and rigor as the medical device itself, and should not be considered secondary to the device when it comes to qualification testing. Ultimately, the packaging is a vessel for the drug, device, or product and needs to contain and protect what is inside.
With an aging and growing global population, emerging markets around the world, and the constant international pressure to reduce healthcare budgets, medical packaging is an area that needs to maintain high standards for quality testing, as well as having the potential for continued design innovation.
To address these challenges, and opportunities, it’s crucial that testing of physical material characterization is conducted with best practices to adhere to international standards; that packaging design is done with the clinician, patient, or other end user in mind; and that data and records management is organized and well maintained to quality standards of the Federal Drug Administration (FDA).
Material Characterization
When many consider what encompasses quality assurance (QA) testing of medical packaging, often times mechanical testing is an obvious and non-negotiable component. For instance, when a manufacturer designs packaging for its medical device, the package integrity is key for keeping what’s inside of the package sterile.
However, it’s equally important that the package is easy to open by the end user, or else it could hinder the intended efforts. Because of this, one of the most common and effective medical packaging standards is ASTM F88-09 for determining the seal strength of flexible barrier packaging.
Measuring seal strength allows packaging labs to obtain quantitative measurements about their packaging process. Often labs report the average force to open the seal and peak load during a test. For this test, it’s critical to set the test speed at an appropriate rate and to ensure the universal testing machine has a high-enough bandwidth to pick up the quick peaks and drops in loads during the peel. Many labs test their medical packaging not only to obtain relevant force data to open the package, but also to measure the consistency of the packaging process and other factors that could affect the strength of the package seal, such as a sterilization process or shelf-time.
In addition to tracking seal strength of a package, many packaging labs also report tensile strength results of the bulk package material. While the package seal is the intended opening location, that’s not always the case. Many consumers tend to poke and puncture the packaging to get a product out. Furthermore, packaging manufacturers need to avoid unintentional package damage caused in shipping, handling, or storage of a product.
For these reasons, medical packaging labs also test to ASTM D882-12 for determining tensile properties of thin plastic sheeting, which is often what flexible packaging consists of. Properties, such as tensile modulus, load at break, and tensile energy at break, are all used to characterize material properties best suited for housing a medical device.
While the same universal testing machine can be used to test seal strength and to test to ASTM D882-12, the tensile test is much faster than the seal strength test, so it’s critical for packaging labs to set the data acquisition of their machines accordingly. If the data acquisition rate is set too low, generally above 100 ms, often tensile modulus results will be calculated from too few data points, resulting in poor stiffness data.
As mentioned, many medical devices are sealed into their sterile packaging in a cleanroom and loaded into protective packaging, or shipping cartons, for transportation. This helps to ensure that the product is supplied to the customer in a sterile state. The transit simulation is carried out on populated shipping cartons, normally with two goals in mind: (1) to assess the performance of the shipping carton and its robustness in transport and (2) to subject the shipping carton’s contents to the rigors of transport (this is more important for the medical device manufacturer). This allows performance testing of the sterile barrier packaging and assessment of the product. A typical example is the concentrated impact test, which simulates the effect of direct shock impact on a package by a concentrated external force, such as when packages collide.
ASTM D6344-09 is intended to evaluate the ability of packaging to resist the force of concentrated impacts from outside sources, such as those encountered in various modes of transportation. This test method determines the ability of packaging to protect contents from such impacts and to evaluate if there is sufficient clearance and/or support between the package itself and its contents. It’s conducted using a free-fall tup (or striker) to test completely filled transport packages for resistance against concentrated low-level impacts, typical of those encountered in the distribution environment. The test result is a pass/fail determination and a record of the energy dissipated by the filled transport package during a low-level concentrated impact.
Usability Testing
Quality testing for packaging is often conducted to abide by ASTM or ISO standards for material characterization, but also can focus on usability testing, especially in the pharmaceutical industry. Pharmaceutical packaging has seen many changes in the last decade, especially in the United States with the aging baby boomer population and with more consumers requiring easy-to-use packaging for self-medication.
At many pharmaceutical companies, medical packaging is not just about flexible packaging, but also includes rigid plastic or glass bottles, droppers, and syringes. Standards exist for testing packaging to ensure batch- to-batch package variation remains low and to provide data on the product’s usability.
ISO 7886-1 sets guidelines for sterile hypodermic single-use syringes for manual use, and specifies the test conditions for aspiration and ejection of liquids. The purpose of the test is to ensure that the forces necessary to move the syringe plunger and eject the fluid from the barrel are not too high or too low. Parameters that affect the amount of force to eject a drug could be the syringe material, viscosity of the liquid, the sterilization process, and geometry of the syringe. In addition to this, the age and gender of the host tissue could also affect the forces required to eject the drug.
When performing QA usability testing on syringes, packaging labs need to focus on the force required to move the plunger (known as the break-away force), as well as the friction of the plunger as it moves through the shaft of the syringe (known as the glide force).
Similarly, in 2014, ASTM Committee D10 on packaging developed ASTM D7860-14, a method for determining torque retention for child-resistant and non-child-resistant packages with continuous thread closures, which can be specifically applied to medicine bottles. Child-resistant medicine packaging is needed to reduce the risk of children ingesting dangerous amounts of medication. However, making packaging too difficult to open by requiring too much force to push down and too much torque to twist may prevent elderly individuals with arthritis or disabled individuals from taking their needed medications.
In addition to safety concerns with opening the packaging, there are also concerns with the packaging of medicine bottles being too difficult to close. Capped medicine bottles that are stored incorrectly—ones that require too much torque to fully close—could lead to a change in dose concentrations of the medicine and cause harmful effects to the consumer. This type of mechanical testing for quality control purposes of medical packaging is not only useful to ensure a uniform manufacturing process, but also to ensure the product has been optimized for ease of use and safety.
Data & Records Management
It’s normal practice for many biomedical, pharmaceutical, and medical device companies to interface with the FDA and their requirements, which are considered throughout the device, drug, or product lifecycle. While a premarket approval may not be needed for a medical device’s packaging, packaging standards and records management should be taken seriously. Testing physical properties of medical packaging for a variety of parameters, and how accurate and reliable those results are, is all important information needed by the FDA.
This is where installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ) to FDA 21 CFR part 820.76 are needed to validate the universal testing machine that’s used to test the medical packaging. Many companies establish their own IQ/OQ/PQ process in their quality departments or require the mechanical testing equipment supplier to help support the process.
The purpose of the IQ/OQ/PQ process is to ensure the testing equipment being used is suitable for its purpose and is capable of producing valid results. The IQ process is designed to ensure that the system is installed and set up correctly. In addition, this process requires the recording of the installation conditions, operation of safety features, environmental conditions, and all appropriate services and utilities of the equipment. While the IQ process is perhaps the simplest part of IQ/OQ/PQ, it’s good documentation practice to have it complete and readily available.
The OQ process verifies the correct operation of the testing system and validates the test results. For medical packaging labs, a validation plan could include system operation validation, verification of the force transducers, functionality checks of the software, and validation of key calculations, such as peak load and tear resistance.
PQ is the most complex part of the validation process. For a medical packaging laboratory, the PQ process validates different system configurations for different test types (e.g., peel test and tensile test), and ensures that different operators use the same protocol to achieve similar results.
After test methods to appropriate ASTM standards are created, protocols are established, and IQ/OQ/PQ validation is complete, a medical packaging QA lab is ready to begin testing. Once quality testing on medical packaging begins, the results will need to be analyzed and documentation will need to be kept organized as to FDA 21 CFR part 11. This FDA standard states that all changes, additions, modifications, and deletions to test data, a testing method, or test file must be recorded.
Today, the majority of medical device companies meet this requirement through secure software that captures all electronic records of a testing system. This becomes applicable for packaging labs when an issue could arise in the field with regards to the packaging of a medical device or drug becoming compromised. Under an FDA audit, the packaging lab would be able to pull an electronic record of all testing results, raw data, and methods along with electronic signatures to produce an audit trail.
Overall, quality testing of medical packaging needs to be evaluated with the same thoroughness as the medical device itself. It’s crucial that testing of physical material characterization is conducted with best practices to adhere to international standards; that packaging is designed with the clinician, patient, or consumer in mind; and that data and records management is organized and well maintained to the quality standards of the FDA. As our global population continues to expand, and pressure to reduce healthcare budgets continuously rises, the medical packaging industry needs to maintain high standards to ensure that safe, easy-to-use, and effective medical products come to the market.

About the author…
Elayne Schneebacher has been with Instron as an applications engineer since 2012 and works specifically with Instron’s Electromechanical business. Prior to Instron, she studied biomedical engineering at Rutgers University with a focus on materials science and tissue engineering. She can be reached at U.S. 781-575-5455 or Elayne_schneebacher@instron.com.