The Potential Role of Anti-PCSK9 Monoclonal Antibodies in the Management of Hypercholesterolemia
Norman E. Lepor, MD, FACC, FAHA, FSCAI,1,2 Laurn Contreras, BS,2 Chirag Desai, MD,2 Dean J. Kereiakes, MD, FACC, FSCAI3
1The David Geffen School of Medicine at UCLA and Cedars-Sinai Medical Center, Los Angeles, CA; 2Westside Medical Associates of Los Angeles, Beverly Hills, CA; 3The Christ Hospital Heart and Vascular Center, The Lindner Research Center, Cincinnati, OH
Atherosclerotic cardiovascular disease (ASCVD) is the leading cause of death and disability in developed nations, and it is rising rapidly in other parts of the developing world. Levels of low-density lipoprotein cholesterol (LDL-C) are directly correlated with atherogenic risk, and statin-based therapy is the most common management for these patients. However, many patients exhibit resistance to and/or adverse effects from statin therapy, and there is a need for adjunctive therapies or statin alternatives for these patients. The recently discovered human protein proprotein convertase subtilisin/ kexin type 9 (PCSK9) plays an important role in LDL-C metabolism. PCSK9 promotes LDL receptor (LDL-R) degradation with a consequent reduction in LDL-R density and an increase in LDL-C levels. Consequently, PCSK9 inhibition to reduce LDL-C levels has become a primary focus for drug development. Numerous clinical trials focusing on monoclonal antibodies against PCSK9 have demonstrated efficacy equal to or greater than statin therapy for lowering LDL-C levels. Long-term trials are underway to assess safety, tolerability, and ability to reduce ASCVD.
[Rev Cardiovasc Med. 2014;15(4):290-309 doi: 10.3909/ricm0773]
© 2015 MedReviews®, LLC
The Potential Role of Anti-PCSK9 Monoclonal Antibodies in the Management of Hypercholesterolemia
Norman E. Lepor, MD, FACC, FAHA, FSCAI,1,2 Laurn Contreras, BS,2 Chirag Desai, MD,2 Dean J. Kereiakes, MD, FACC, FSCAI3
1The David Geffen School of Medicine at UCLA and Cedars-Sinai Medical Center, Los Angeles, CA; 2Westside Medical Associates of Los Angeles, Beverly Hills, CA; 3The Christ Hospital Heart and Vascular Center, The Lindner Research Center, Cincinnati, OH
Atherosclerotic cardiovascular disease (ASCVD) is the leading cause of death and disability in developed nations, and it is rising rapidly in other parts of the developing world. Levels of low-density lipoprotein cholesterol (LDL-C) are directly correlated with atherogenic risk, and statin-based therapy is the most common management for these patients. However, many patients exhibit resistance to and/or adverse effects from statin therapy, and there is a need for adjunctive therapies or statin alternatives for these patients. The recently discovered human protein proprotein convertase subtilisin/ kexin type 9 (PCSK9) plays an important role in LDL-C metabolism. PCSK9 promotes LDL receptor (LDL-R) degradation with a consequent reduction in LDL-R density and an increase in LDL-C levels. Consequently, PCSK9 inhibition to reduce LDL-C levels has become a primary focus for drug development. Numerous clinical trials focusing on monoclonal antibodies against PCSK9 have demonstrated efficacy equal to or greater than statin therapy for lowering LDL-C levels. Long-term trials are underway to assess safety, tolerability, and ability to reduce ASCVD.
[Rev Cardiovasc Med. 2014;15(4):290-309 doi: 10.3909/ricm0773]
© 2015 MedReviews®, LLC
The Potential Role of Anti-PCSK9 Monoclonal Antibodies in the Management of Hypercholesterolemia
Norman E. Lepor, MD, FACC, FAHA, FSCAI,1,2 Laurn Contreras, BS,2 Chirag Desai, MD,2 Dean J. Kereiakes, MD, FACC, FSCAI3
1The David Geffen School of Medicine at UCLA and Cedars-Sinai Medical Center, Los Angeles, CA; 2Westside Medical Associates of Los Angeles, Beverly Hills, CA; 3The Christ Hospital Heart and Vascular Center, The Lindner Research Center, Cincinnati, OH
Atherosclerotic cardiovascular disease (ASCVD) is the leading cause of death and disability in developed nations, and it is rising rapidly in other parts of the developing world. Levels of low-density lipoprotein cholesterol (LDL-C) are directly correlated with atherogenic risk, and statin-based therapy is the most common management for these patients. However, many patients exhibit resistance to and/or adverse effects from statin therapy, and there is a need for adjunctive therapies or statin alternatives for these patients. The recently discovered human protein proprotein convertase subtilisin/ kexin type 9 (PCSK9) plays an important role in LDL-C metabolism. PCSK9 promotes LDL receptor (LDL-R) degradation with a consequent reduction in LDL-R density and an increase in LDL-C levels. Consequently, PCSK9 inhibition to reduce LDL-C levels has become a primary focus for drug development. Numerous clinical trials focusing on monoclonal antibodies against PCSK9 have demonstrated efficacy equal to or greater than statin therapy for lowering LDL-C levels. Long-term trials are underway to assess safety, tolerability, and ability to reduce ASCVD.
[Rev Cardiovasc Med. 2014;15(4):290-309 doi: 10.3909/ricm0773]
© 2015 MedReviews®, LLC

KEY WORDS
Hypercholesterolemia • PCSK9 • Low-density lipoprotein cholesterol • Atherosclerotic cardiovascular disease • Anti-PCSK9 monoclonal antibodies
KEY WORDS
Hypercholesterolemia • PCSK9 • Low-density lipoprotein cholesterol • Atherosclerotic cardiovascular disease • Anti-PCSK9 monoclonal antibodies
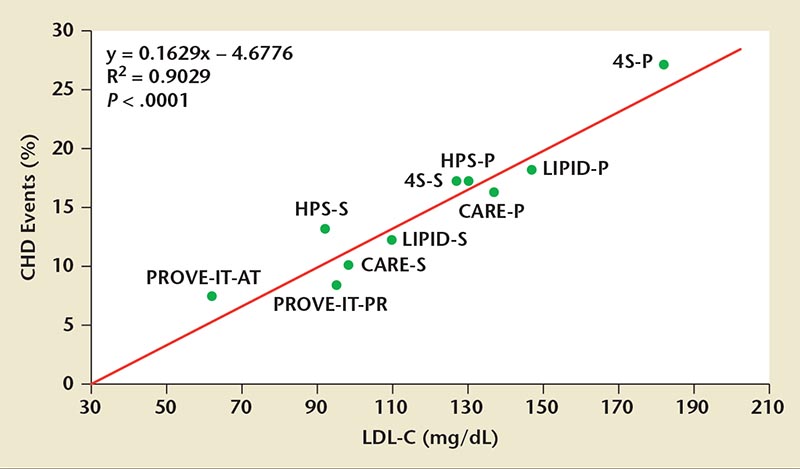
Figure 1. In statin secondary prevention trials, CHD event rates were directly proportional to on-treatment LDL-C levels. 4S, Scandinavian Simvastatin Survival Study; CARE, Cholesterol And Recurrent Events Trial; CHD, coronary heart disease; HPS, Heart Protection Study; LDL-C, low-density lipoprotein cholesterol; LIPID, Longterm Intervention with Pravastatin in Ischemic Disease Trial; PROVE-IT, Pravastatin or Atorvastatin Evolution and Infection Therapy trial. Reprinted with permission from O’Keefe JH Jr et al.9
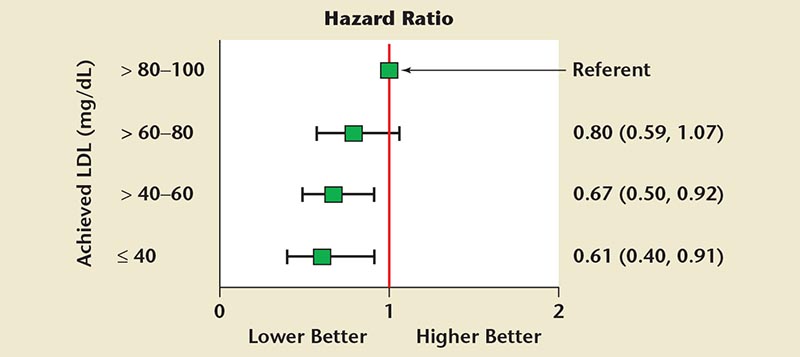
Figure 2. Hazard ratio of atherosclerotic cardiovascular disease composite (primary endpoint) versus range of on-statin low-density lipoprotein cholesterol (LDL-C) level (adjusted for age, sex, baseline LDL-C, diabetes, and prior myocardial infarction). Adapted with permission from Wiviott SD et al.10
Of the general population of patients who have dyslipidemia and coronary risk, an estimated 10% to 20% cannot tolerate either statins or the statin dose required to achieve current LDL-C guideline goals. Adverse side effects from statins are relatively common, limiting or preventing their use in many patients.
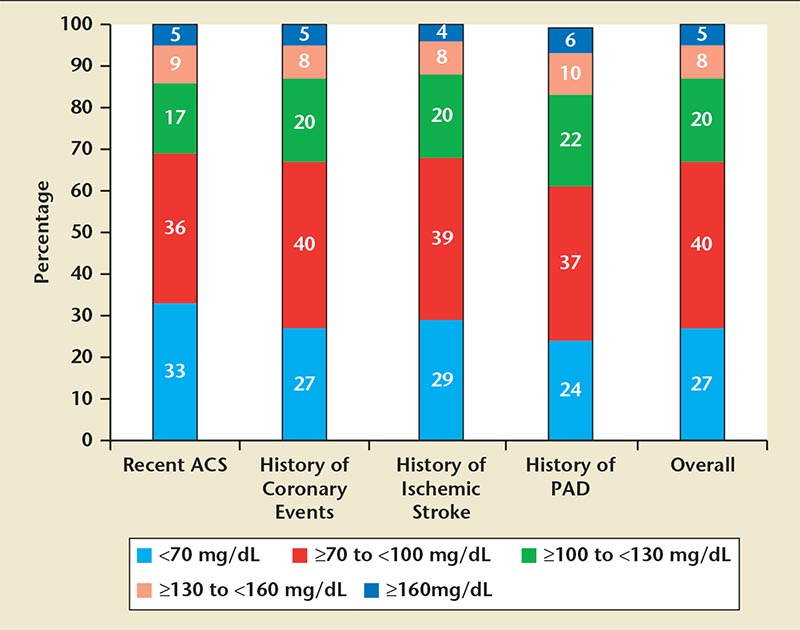
Figure 3. Achieved low-density lipoprotein cholesterol by very high cardiovascular risk categories, including those with recent acute coronary syndrome (ACS), history of coronary events, history of ischemic stroke, and history of peripheral arterial disease (PAD). Data from Ray KK et al. ISPOR 16th Annual European Congress; November 2-6, 2013; Dublin, Ireland.
Sequencing of the PCSK9 gene in individuals with low LDL-C found that those with loss-of-function PCSK9 mutations had a 28% lower LDL-C level compared with individuals with no mutation.
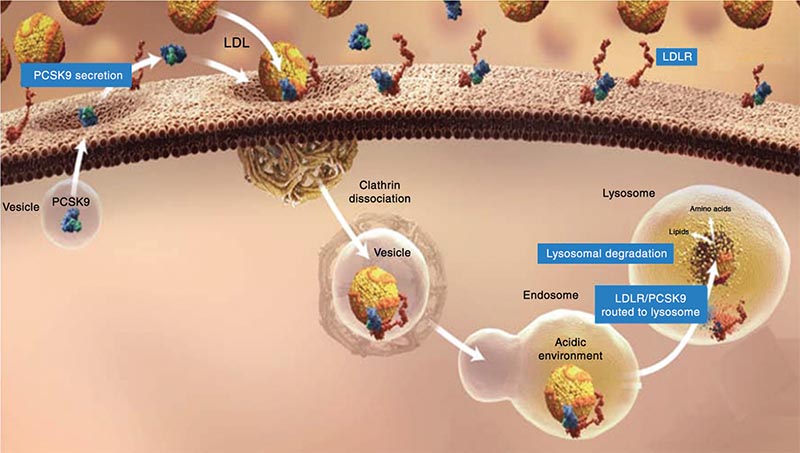
Figure 4. PCSK9 binds to the EGF-A domain of the LDL-R, stimulating LDL-R degradation. When this complex is internalized in clathrin-coated endosomes, the LDL-R bound to PCSK9 undergoes lysosomal degradation. EGF-A, epidermal growth factor-like repeat A; LDL-R, low-density lipoprotein receptor; PCSK9, proprotein convertase subtilisin/kexin type 9. Image from Davidson MH. http://www.medscape.org/viewarticle/763848_transcript. Accessed October 28, 2014.
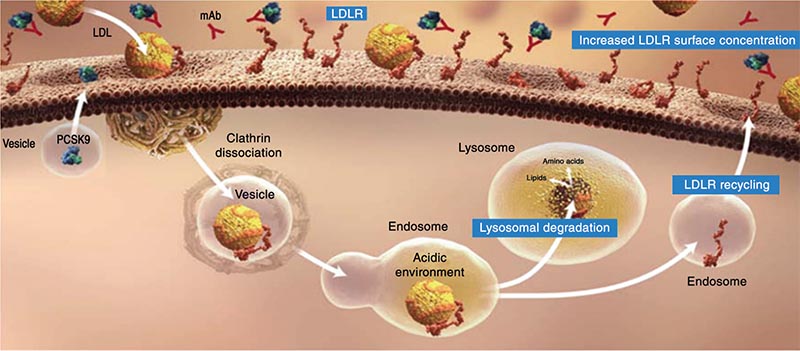
Figure 5. By targeting the LDL-R, PCSK9 reduces its functional half-life and results in reducing the hepatocyte’s ability to clear LDL-C from the circulation. LDL-C, lowdensity lipoprotein cholesterol; LDL-R, low-density lipoprotein receptor; mAb, monoclonal antibody; PCSK9, proprotein convertase subtilisin/kexin type 9. Image from Davidson MH. http://www.medscape.org/viewarticle/763848_transcript. Accessed October 28, 2014.
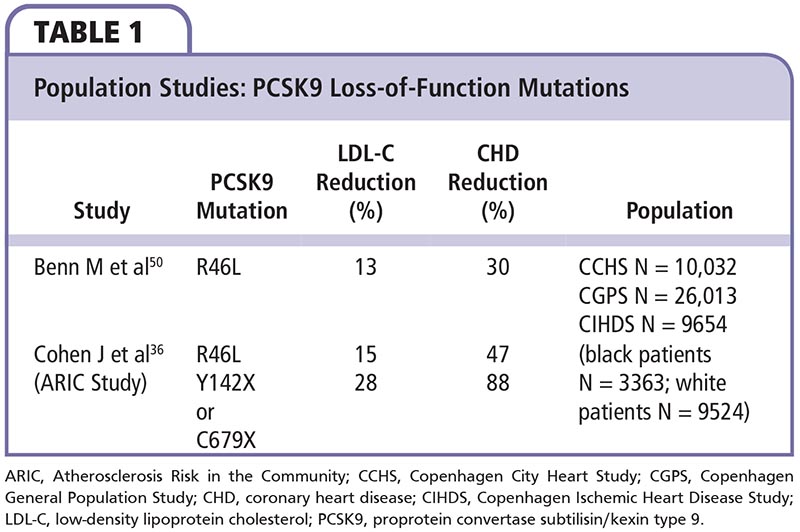
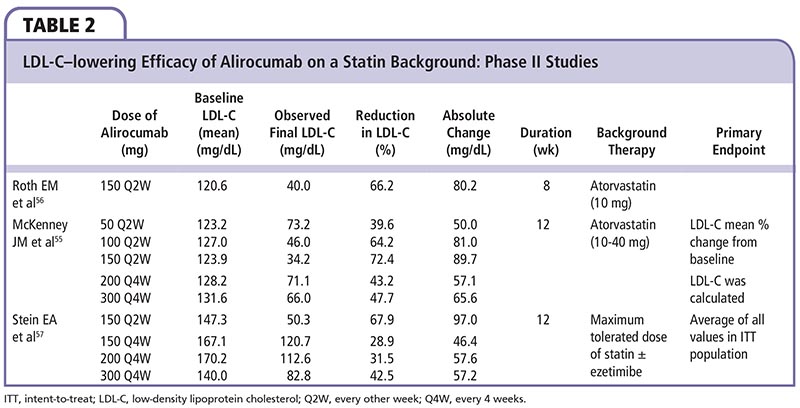
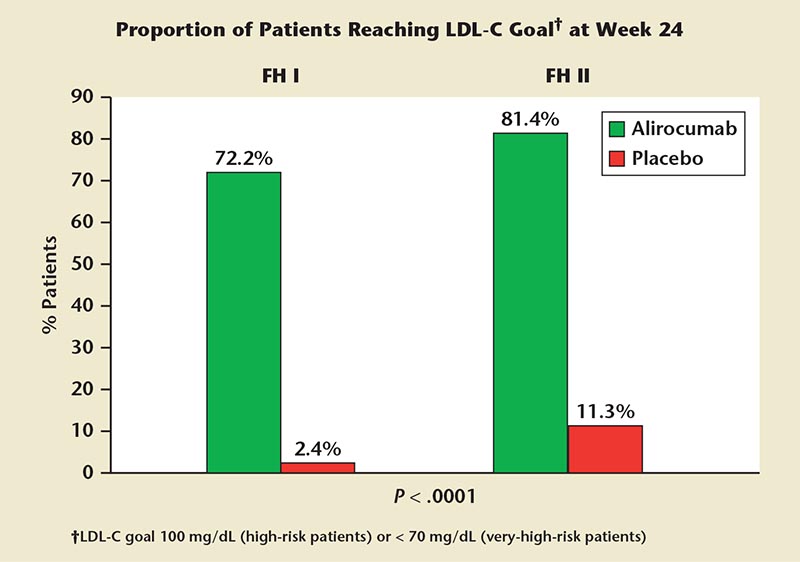
Figure 6. ODYSSEY FH I and FH II. Most heterozygous familial hypercholesterolemia patients receiving alirocumab on background statin ± other lipid-lowering therapy achieved low-density lipoprotein cholesterol (LDL-C) goals. ODYSSEY FH I, Efficacy and Safety of Alirocumab (SAR236553/REGN727) Versus Placebo on Top of Lipid-Modifying Therapy in Patients With Heterozygous Familial Hypercholesterolemia Not Adequately Controlled With Their Lipid-Modifying Therapy); ODYSSEY FH II, Study of Alirocumab (REGN727/ SAR236553) in Patients With HeFH (Heterozygous Familial Hypercholesterolemia) Who Are Not Adequately Controlled With Their LMT [Lipid-Modifying Therapy].
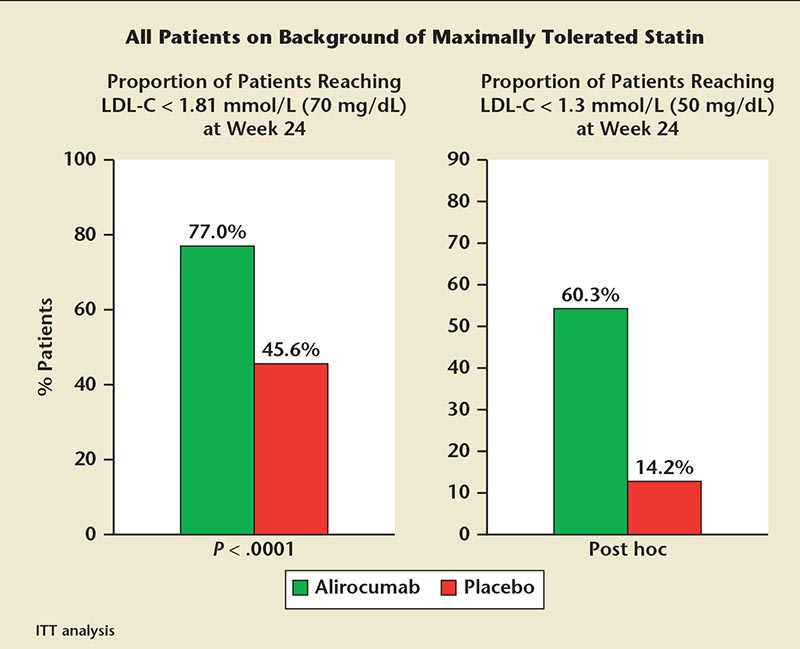
Figure 7. ODYSSEY COMBO II. Most of these high-cardiovascular risk patients receiving alirocumab on background statin achieved LDL-C goal. ITT, intention-to-treat; LDL-C, low-density lipoprotein cholesterol; ODYSSEY COMBO II, Efficacy and Safety of Alirocumab (SAR236553/REGN727) Versus Ezetimibe on Top of Statin in High Cardiovascular Risk Patients With Hypercholesterolemia.
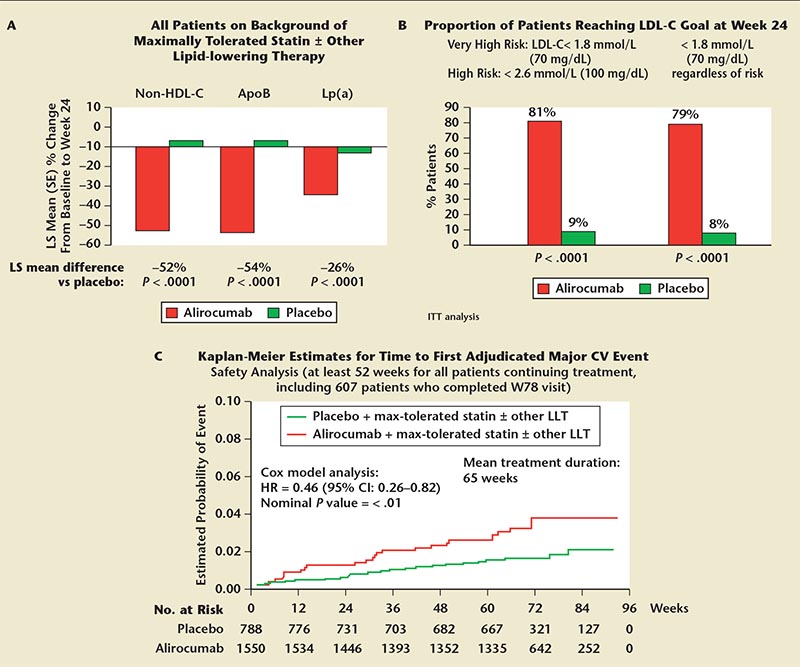
Figure 8. ODYSSEY LONG TERM. (A) Significant reductions in secondary lipid parameters at week 24. Adjusted mean (SE) shown for Lp(a). (B) Most patients receiving alirocumab on background statin ± other LLT achieved LDL-C goals. (C) Post hoc adjudicated cardiovascular treatment-emergent adverse events. Same as primary endpoint of ongoing ODYSSEY OUTCOMES trial: coronary heart disease death, nonfatal myocardial infarction, fatal and nonfatal ischemic stroke, unstable angina requiring hospitalization. ApoB, apolipoprotein B; CI, confidence interval; CV, cardiovascular; HDL-C, high-density lipoprotein cholesterol; HR, hazard ratio; ITT, intention- to-treat; LDL-C, low-density lipoprotein cholesterol; LLT, lipid-lowering therapy; Lp(a), lipoprotein(a); LS, least squares; ODYSSEY Long Term, Long-term Safety and Tolerability of Alirocumab (SAR236553/REGN727) Versus Placebo on Top of Lipid-Modifying Therapy in High Cardiovascular Risk Patients With Hypercholesterolemia; ODYSSEY Outcomes, Evaluation of Cardiovascular Outcomes After an Acute Coronary Syndrome During Treatment With Alirocumab SAR236553 (REGN727); SE, standard error.
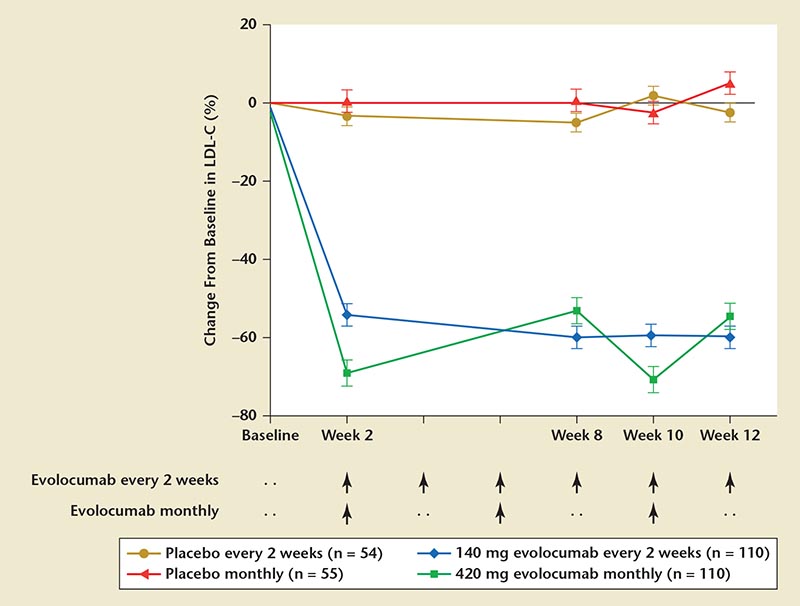
Figure 9. RUTHERFORD-2: change from baseline with evolocumab versus placebo. LDL-C, low-density lipoprotein cholesterol; PCSK9, proprotein convertase subtilisin/kexin type 9; RUTHERFORD, Reduction of LDL-C With PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder. Reprinted with permission from Raal FJ et al.78

Figure 10. MENDEL-2: mean percentage change in LDL-C from baseline, placebo versus ezetimibe. LDL-C, low-density lipoprotein cholesterol; MENDEL, Monoclonal Antibody Against PCSK9 to Reduce Elevated LDL-C in Patients Currently Not Receiving Drug Therapy for Easing Lipid Levels; PCSK9, proprotein convertase subtilisin/ kexin type 9; SC, subcutaneous. Reprinted with permission from Koren MJ et al.79
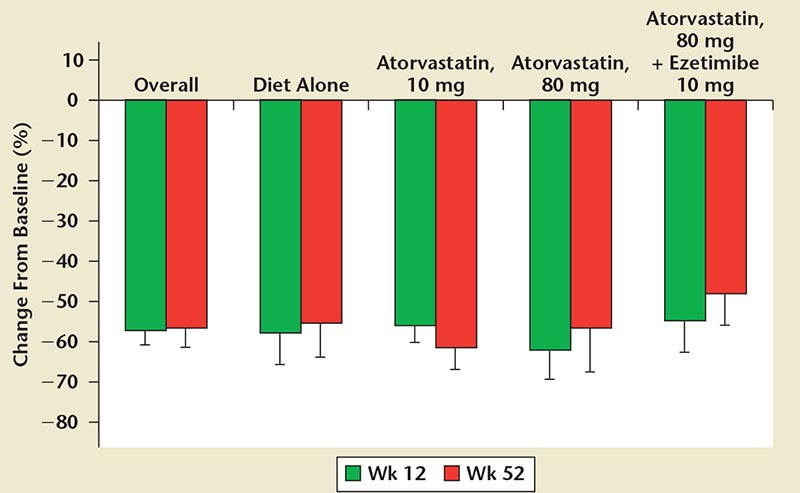
Figure 11. DESCARTES: percentage reduction in mean LDL-C from baseline versus placebo and atorvastatin dosages. DESCARTES, Durable Effect of PCSK9 Antibody Compared with Placebo; LDL-C, low-density lipoprotein cholesterol; PCSK9, proprotein convertase subtilisin/kexin type 9. Reprinted with permission from Blom DJ et al.81
Main Points
• To reduce cardiovascular risk, low-density lipoprotein cholesterol (LDL-C) reduction remains the primary target for lipid modification.
• Many patients are not able to reach LDL-C treatment goals with statins due to intolerance or resistance, particularly those with heterozygous familial hypercholesterolemia.
• Proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors increase the functional half-life of the LDL receptor, leading to increased removal of LDL-C from the circulation. PCSK9 inhibitors are effective either as monotherapy or as an adjunct to other lipid-lowering therapies, reducing LDL-C levels 50% to 70%, and appear to be well tolerated and safe.
• Signals are appearing in clinical trials that suggest a substantial incremental reduction of cardiovascular events with PCSK9 inhibition above and beyond those related to statins.
• Large randomized clinical trials with cardiovascular events as the primary endpoint are in progress: ODYSSEY OUTCOMES, FOURIER, and SPIRE.
Main Points
• To reduce cardiovascular risk, low-density lipoprotein cholesterol (LDL-C) reduction remains the primary target for lipid modification.
• Many patients are not able to reach LDL-C treatment goals with statins due to intolerance or resistance, particularly those with heterozygous familial hypercholesterolemia.
• Proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors increase the functional half-life of the LDL receptor, leading to increased removal of LDL-C from the circulation. PCSK9 inhibitors are effective either as monotherapy or as an adjunct to other lipid-lowering therapies, reducing LDL-C levels 50% to 70%, and appear to be well tolerated and safe.
• Signals are appearing in clinical trials that suggest a substantial incremental reduction of cardiovascular events with PCSK9 inhibition above and beyond those related to statins.
• Large randomized clinical trials with cardiovascular events as the primary endpoint are in progress: ODYSSEY OUTCOMES, FOURIER, and SPIRE.
Atherosclerosis is caused primarily by dyslipidemia and other risk factors that promote cholesterol deposition in the arterial intima.1 In most patients, low-density lipoprotein cholesterol (LDL-C) appears to account for the vast majority of atherogenesis risk. An estimated 57 million US adults age ≥ 20 years (25.3%) have plasma LDL-C levels ≥ 160 mg/dL.2 Atherosclerotic cardiovascular disease (ASCVD) is the leading cause of death and disability in the United States and developed nations, and prevalence is rising rapidly in other parts of the developing world.
The Framingham Heart Study was the first to show a correlation between elevated total cholesterol and LDL-C with ASCVD-related events.3 Results from numerous randomized clinical trials (RCTs) of both statin and nonstatin treatments over several decades have also shown a reduced ASCVD incidence with therapies that lower LDL-C.4-8 Specifically, results have shown elevated LDL-C levels are causally related to atherosclerosis and lowering LDL-C levels will decrease ASCVD events and mortality in proportion to the degree of LDL-C lowering.4-9
Statins are currently the most effective commercially available agents for reducing LDL-C levels; an estimated 20 million patients have been prescribed this treatment. In statin secondary prevention trials, coronary heart disease (CHD) event rates were directly proportional to LDL-C levels obtained during treatment, demonstrating a causal link to atherosclerosis risk (Figure 1). ASCVD risk declines progressively with lower “on-statin treatment” LDL-C levels (Figure 2).10
Epidemiologic data have shown an inverse relationship between high-density lipoprotein cholesterol (HDL-C) levels, as well as a direct relationship between triglyceride levels and ASCVD risk. Cardioprotective effects of HDL-C have been attributed to several factors,11 including reverse cholesterol transport, effects on endothelial cell function, and antioxidant activity. However, clinical trial outcomes evaluating HDL-C-targeted therapies, including niacin, fibric acid derivatives, and cholesteryl ester transfer protein (CETP) inhibitors, have lacked strength and consistency in comparison with LDL-C reduction.11 The ongoing Reduction of Cardiovascular Events with EPA-Intervention (REDUCE-IT) trial is evaluating the omega-3 fish oil icosapent ethyl and its impact on cardiovascular (CV) events.12
Evidence of Need: Looking Beyond Statins
Heterozygous Familial Hypercholesterolemia
Heterozygous familial hypercholesterolemia (HeFH) is one of the most common genetic diseases, with a prevalence of 1 in 200 to 1 in 500 individuals,13 and is characterized by extremely high levels of LDL-C, premature atherosclerosis, and CV disease.14 A diagnosis of HeFH is made in only 1% of afflicted patients, and many are not identified until they have had a cardiac event. Through careful clinical observation and genetic analysis, individuals with HeFH can be identified. Diagnosis in the clinical setting begins with obtaining a full individual and family medical history. A strong family history of elevated and therapy-resistant total cholesterol and LDL-C levels in one parent and early onset of CHD or premature myocardial infarction (MI) should raise suspicion of a HeFH.13,15 An individual medical history of HeFH is likely to include elevated total and LDL-C levels, premature CHD, physical evidence of cholesterol deposits, or a history of Achilles tendonitis.16
Lipid analysis with the presence of severe LDL-C elevation (LDL-C > 250 mg/dL in adults or LDL-C > 200 mg/dL in individuals age < 20 years) and severe elevation of total cholesterol (> 310 mg/dL in adults or > 230 mg/dL in individu als age < 20 years) in the absence of secondary causes of hyperlipidemia can further indicate pos sible HeFH.13,17,18 Lipid analysis in individuals with HeFH will reveal relatively normal to slightly elevated triglyceride levels. Should an elevated triglyceride level be present in conjunction with both elevated total and LDL-C, diagnosis of HeFH should be approached with caution.19
Further diagnosis can be accomplished through physical examination with identification of cholesterol deposits on the skin and tendons (xanthomas), eyelids (xanthelasmas), and/or cornea of the eye (corneal arcus). Tendon xanthomas occur in approximately 40% of HeFH patients over age 40 years,19 and appear to be isolated to the Achilles tendon and metacarpal phalangeal extensor tendons of the hand. Because xanthomas may not be apparent by visual inspection, palpitation will reveal diffusely thickened tendons suggestive of xanthomas. Tendon xanthomas of the metacarpal phalangeal joints may seem more apparent by visual inspection with flexing of the digits to observe nodules that move with the tendon motion, thereby distinguishing xanthomas from existing subcutaneous nodules.15,19 Xanthomas may be imaged using sonography for further confirmation. Other studies that evaluate heart function or cardiac stress testing may yield abnormal results, suggestive of premature ischemic heart disease.15
Clinical parameters for identification of HeFH require genetic analysis for a definitive diagnosis, so that other genetic mutations, such as familial ligand defective apolipoprotein B-100 that share the physical manifestations and elevated lipid measurements of HeFH, are excluded.20 Molecular testing will demonstrate a pathogenic defect in the LDLR or APOB genes.20 The presence of a functional mutation in one copy of the LDLR gene yields a definitive diagnosis of HeFH.
Despite aggressive lipid-lowering therapy (LLT), many patients with HeHF still have LDL-C > 100 mg/dL, and even more have LDL-C > 70 mg/dL.13,14 Statin therapy is often insufficient for these patients to achieve LDL-C treatment goals.21
Statin Intolerance
Of the general population of patients who have dyslipidemia and coronary risk, an estimated 10% to 20% cannot tolerate either statins or the statin dose required to achieve current LDL-C guideline goals. Adverse side effects from statins are relatively common,21 limiting or preventing their use in many patients. The proportion of ASCVD patients treated with high-potency statins increased only from 13% in 2004 to 26% in 2012.22 Even at current guideline-recommended statin use, CV risk reduction with statin monotherapy is between 25% and 50%; therefore, most ASCVD events are not prevented.23 Indeed, residual ASCVD risk is, in large part, related to the level of residual dyslipidemia as reflected by LDL-C or non-HDL-C level (combination of LDL-C and very low-density lipoprotein levels).
Overcoming Statin Resistance
Insufficient control of LDL-C and non-HDL-C levels may be exacerbated by diabetes, metabolic syndrome, or other factors that elevate ASCVD risk and/or non-HDL-C, such as HeFH, or in individuals with other genetic defects (eg, single nucleotide polymorphisms at SORT1/CELSR2/PSRC1 and variants at the APOE and LPA loci).24-26
Most currently available non-statin lipid medications, including niacin, fibrates, and CETP inhibitors, reduce LDL-C by 10% to 20%. Although these therapies are compatible with statins, they have not been associated with significant reductions in ASCVD events in previous clinical trials. Ezetimibe, which inhibits the Niemann-pick C1-like 1 protein, functions as a sterol transporter to mediate intestinal cholesterol absorption and counterbalances hepatobiliary cholesterol excretion. When added to 40 mg of simvastatin in the Improved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) study for patients with a recent acute coronary syndrome (ACS), ezetimibe reduced the composite CV event rate (CV death, MI, documented unstable angina requiring hospitalization, coronary revascularization > 30 days, or stroke) by 6.4% (P = .016).27,28 This represents a modest reduction of CV events corresponding to the modest additional reduction of LDL-C obtained with ezetimibe. The 2013 American College of Cardiology/American Heart Association (ACC/AHA)cholesterol guidelines statement that no statin adjuncts have a favorable risk-to-benefit ratio will need to be revised.29 These data are consistent with the premise that “lower is better” with respect to LDL-C.
Ray and colleagues30 reviewed the primary findings of a British survey investigating an LDL-C treatment goal of < 70 mg/dL in high- and very high-CV risk patients (Figure 3). High-risk CV patients were identified as those with recent ACS, or a history of prior coronary events, ischemic stroke, or peripheral artery disease. Overall, 27% of patients from The Health Improvement Network database achieved LDL-C levels < 70 mg/dL; an additional 40% achieved LDL-C levels between 70 and ≥ 100 mg/dL, and the final third achieved LDL-C levels > 100 mg/dL.30 The LDL-C findings were similar across all listed types of high-risk CV conditions. Approximately two-thirds of patients with a condition indicating very-high CV risk (eg, ACS) did not reach the treatment target LDL-C goal of < 70 mg/dL. These data emphasize the need for ongoing efforts to improve prescription of appropriate additional LLTs and to identify new therapies for improving LDL-C goal attainment, particularly among high-risk patients.
PCSK9 Protein
Proprotein convertase substilisin/ kexin type 9 (PCSK9) is a naturally occurring protein that targets the LDL receptor (LDL-R) for degradation by creating a PCSK9-LDL-R receptor complex.31,32 The LDL-R located on the hepatocyte cell surface mediates LDL-C endocytosis and all atherogenic lipoproteins by recognizing apolipoproteins B and E. PCSK9 is a key determinant of hepatic LDL-R density through its effect on LDL-R catabolism.31,32 PCSK9 promotes lysosomal degradation of intracellular LDL-R and, thus, prevents LDL-R recycling with a consequent reduction in hepatocellular surface membrane LDL-R density.
Function and Physiology
The role of PCSK9 in modulating lipid metabolism was first identified in 2003.33 A case of autosomal-dominant familial hypercholesterolemia was defined in relation to the gene encoding PCSK9. Additional studies in genetically modified animals (knock-out model) demonstrated that PCSK9 loss of function resulted in decreased LDL-C levels.34 Sequencing of the PCSK9 gene in individuals with low LDL-C found that those with loss-of-function PCSK9 mutations had a 28% lower LDL-C level compared with individuals with no mutation.35,36 A relative 88% decrease in ASCVD events was observed in 15-year follow-up in these patients.36 Gain-of-function mutations in PCSK9 reduced hepatic LDL-C receptors and resulted in higher plasma LDL-C levels.33 PCSK9 mRNA levels are responsive to cellular cholesterol levels through the transcription factor sterol regulatory element-binding protein-2 (SREBP-2).37 Furthermore, statins also up-regulate PCSK9 synthesis, which may counterbalance their primary objective to increase LDL-R.38
PCSK9 binds to the epidermal growth factor-like repeat A domain of the LDL-R, stimulating LDL-R degradation (Figure 4).32,39,40 When this complex is internalized in the clathrin-coated endosomes, the LDL-R bound to PCSK9 undergoes lysosomal degradation.39,41 By targeting the LDL-R, PCSK9 reduces receptor functional half-life (Figure 5) and, therefore, hepatocellular LDL-C clearance from the circulation with consequent elevation in LDL-C levels.39 This process is, in large part, related to SREBP-2.42 Up-regulation of SREBP-2 occurs in response to low cholesterol levels in key intra-hepatic regulatory pools, as well as to statins and other LLTs.42,43 Increased uptake of LDL-C from the plasma and PCSK9 transcription leads to increased LDL-R degradation. Adding a therapy to inhibit PCSK9 binding to the LDL-R is an intuitively attractive option to lower LDL-C levels.44
PCSK9 Inhibition Process
Monoclonal antibody (mAb)-mediated mechanisms have been the primary focus for PCSK9 inhibition,45 and have demonstrated greater efficacy for lowering LDL-C than statins in clinical trials. These mAbs appear to be well tolerated and safe in clinical trial experience to date. Non-antibody PCSK9 inhibition compounds also exist.46,47 These include adnectins, therapeutic proteins that bind to targets with very high affinity and specificity (similar to antibodies) but are easier to genetically manipulate,45 and antisense oligonucleotides (small interfering RNA), compounds that inhibit PCSK9 gene expression and synthesis.46
The PCSK9 mutation was originally found in autosomal-dominant HeFH individuals, and identified as the gene coding for PCSK9.33,48,49 Loss of PCSK9 function resulted in a relatively modest 15% to 28% decrease in plasma LDL-C, but a striking 47% to 88% reduction in CHD events.36 This greater degree of CV event reduction than anticipated was explained, in part, by a lifetime exposure to lower LDL-C levels.
Two population studies36,50 have shown the beneficial effects of loss-of-function PCSK9 mutations on LDL-C levels and CV outcomes (Table 1). Individuals with loss-of-function mutations in PCSK9 or total lack of PCSK9 have naturally low LDL-C levels and a reduced risk of CHD. These patients are also generally healthy with no other apparent metabolic abnormalities.36,50,51
PCSK9 Inhibitor Clinical Trials
A number of PCSK9 mAb pharmaceuticals are in development, with the greatest clinical trial data available for evolocumab (AMG 145; Amgen, Thousand Oaks, CA),52 alirocumab (REGN727; sanofiaventis, Bridgewater, NJ, and Regeneron, Tarrytown, NY),53 and bococizumab (RN316; Pfizer, New York, NY).54
Alirocumab Clinical Trials
Several phase II clinical trials55-57 have demonstrated LDL-C-lowering efficacy of PCSK9 mAb inhibitors (Table 2) with relatively limited and mild treatment-associated adverse effects (eg, injection site reactions, diarrhea, fatigue, and headache).
McKenney and colleagues55 evaluated the LDL-C-lowering efficacy of alirocumab versus placebo following 12 weeks of therapy in patients with LDL-C ≥ 100 mg/dL who were receiving stable atorvastatin therapy of 10 to 40 mg/d. When added to atorvastatin, alirocumab further reduced LDL-C by 40% to 72% when administered in doses of 50 mg or 150 mg every 2 weeks, respectively.
Roth and associates56 evaluated 92 patients who had LDL-C levels ≥ 100 mg/dL after treatment with atorvastatin, 10 mg, for at least 7 weeks in a phase II, multicenter, double-blind, placebo-controlled trial. The patients were randomly assigned to receive 8 weeks of treatment with atorvastatin, 80 mg/d, plus alirocumab once every 2 weeks, atorvastatin, 10 mg/d, plus alirocumab once every 2 weeks, or atorvastatin, 80 mg/d, plus placebo once every 2 weeks. The least-squares mean (± standard error) percentage reduction from baseline for LDL-C was 73.2 ± 3.5% with atorvastatin, 80 mg, plus alirocumab versus 17.3 ± 3.5% with atorvastatin, 80 mg, plus placebo (P < .001) and 66.2 ± 3.5% with atorvastatin, 10 mg, plus alirocumab.
Stein and associates57 assessed efficacy and safety of various alirocumab doses and dosing intervals added to maximally tolerated statins with or without ezetimibe to lower LDL-C in HeFH patients. Mean LDL-C reduction from baseline to week 12 was 28.9% with alirocumab, 150 mg, every 4 weeks (P = .0113), 31.54% with alirocumab, 200 mg, every 4 weeks (P = .0035), 42.53% with alirocumab, 300 mg, every 4 weeks (P < .0001), and 67.90% with alirocumab, 150 mg, every 2 weeks (P < .0001) versus 10.65% for placebo. Alirocumab was well tolerated and achieved a substantial further reduction in LDL-C among HeFH patients with elevated LDL-C treated with high-dose statins with or without ezetimibe, thus providing more optimal LDL-C control in these difficult-to-treat HeFH patients.
Alirocumab Key Phase III Trials: The ODYSSEY Trial Program
The phase III ODYSSEY trial program includes 14 global trials with more than 23,500 patients enrolled across more than 2000 study centers. The trial components include ODYSSEY FH I, II, and HIGH FH; ODYSSEY MONO; ODYSSEY COMBO I and II; ODYSSEY LONG TERM; ODYSSEY CHOICE I and II; ODYSSEY ALTERNATIVES; ODYSSEY OPTIONS I and II; ODYSSEY OUTCOMES; and ODYSSEY OLE.
The ODYSSEY FH I, II, and HIGH FH trials assessed efficacy and safety in HeFH subjects with treatment target goals of LDL-C level 100 mg/dL (high-risk patients) or < 70 mg/dL (very high-risk patients).58,59 Alirocumab dosed at 75 or 150 mg every 2 weeks significantly reduced LDL-C from baseline to week 24 (48.8% [ODYSSEY FH I] and 48.7% [ODYSSEY FH II], respectively) compared with an increase in the placebo arms (9.1% [ODYSSEY FH I] and 2.8% [ODYSSEY FH II], respectively; P < .0001 for all comparisons).59-61 By week 24 (Figure 6), more alirocumab-treated patients (vs placebo-treated patients) reached LDL-C treatment goals (72.2% vs 2.4% in ODYSSEY FH I, and 81.4% vs 11.3% in ODYSSEY FH II; both P < .0001). The mean achieved LDL-C levels were 65.9 mg/dL (ODYSSEY FH II) and 74.3 mg/dL (ODYSSEY FH I) at week 52 with alirocumab.60,61 Pooled data showed that adverse events occurred with a similar incidence in the alirocumab- and placebo-treated groups (74.8% vs 75.4%). Final results from the ODYSSEY HIGH FH clinical trial showed a reduction of LDL-C of 46% with alirocumab versus 6.6% in the placebo group in patients on maximally tolerated statin plus other LLT.58
The ODYSSEY MONO trial assessed efficacy and safety of alirocumab as monotherapy versus ezetimibe in patients not receiving statins or other LLT.59,62 Mean LDL-C reductions at week 24 were 47.3% with alirocumab versus 15.6% with ezetimibe (P < .0001) by intent-to-treat analysis and 54.2% versus 17.2%, respectively, by on-treatment analysis (P < .0001).62 Adverse events were comparable between treatment groups.
The ODYSSEY COMBO I and II trials evaluated the efficacy and safety of alirocumab as add-on therapy to stable, maximally tolerated daily statin therapy versus ezetimibe in patients with hyper-cholesterolemia at high risk of ASCVD.59,63,64 In the ODYSSEY COMBO II trial, alirocumab lowered LDL-C levels significantly more than ezetimibe by week 24 (50.6% vs 20.7%; P < .0001) and week 52 (49.5% vs 18.3%; P < .001) (Figure 7).59,64 In addition, 77% of alirocumab-treated patients achieved LDL-C levels of ≥ 70 mg/dL by week 24 versus 45.6% of ezetimibe-treated patients (P < .0001).59,64 The percentage of adverse events was similar between alirocumab (71.2%) and ezetimibe (67.2%) treatments.59 ODYSSEY COMBO I evaluated LDL-C in high-CV risk subjects on maximally tolerated statin therapy with or without other LLT. Eligible subjects were randomly assigned to treatment with either alirocumab, 75 mg, every 2 weeks or placebo. If LDL-C was > 70 mg/dL at week 8, study drug dose was increased to 150 mg every 2 weeks. ODYSSEY COMBO I demonstrated that (1) alirocumab reduced LDL-C from baseline to week 24 by 48% (vs 2% placebo; P < .0001) and LDL-C reduction was maintained through 52 weeks of therapy; (2) a “treat-to-target” approach resulted in 83% of subjects not requiring a dose increase to 150 mg and almost 80% achieving target LDL-C < 70 mg/dL at week 24; (3) the incidence of adverse events was similar between alirocumab and placebo.65
The ODYSSEY LONG TERM trial assessed the long-term safety and tolerability of alirocumab for the treatment of high- and very high-CV risk patients with HeFH and ASCVD who were not adequately controlled on lipid-modifying therapy.59 By 24 weeks of treatment (Figure 8A), mean LDL-C was reduced from baseline by 61% for alirocumab versus an increase of 0.8% for patients treated with placebo (P < .0001).66 By week 24 (Figure 8B), more alirocumab-treated patients (vs placebo-treated patients) reached LDL-C goals of ≥ 50% reduction frombaseline(76%vs2%;P<.0001); LDL-C level < 100 mg/dL in high-risk patients or < 70 mg/dL in very high-risk patients (81% vs 9%; P < .0001).66 Safety analysis showed that adverse events were similar in both treatment arms (78.6% alirocumab and 80.6% placebo).59,66 A post-hoc analysis of ODYSSEY LONG TERM (Figure 8C) showed a lower rate of major CV events (cardiac death, MI, stroke, and unstable angina requiring hospi-talization) in the alirocumab arm versus placebo (54% reduction over 65 weeks; P < .01).59,66
ODYSSEY CHOICE I and II are ongoing double-blind, placebo-controlled RCTs (duration, 12 months) evaluating alirocumab in two dosages (75 mg every 2 weeks and 300 mg every 4 weeks) versus placebo with a 2:1:4 randomization.59,67,68 The trial objectives are to demonstrate LDL-C reduction with alirocumab at a starting dose of 150 mg every 4 weeks as add-on to nonstatin lipid-modifying background therapy or when administered as monotherapy versus placebo in patients with primary hypercholesterolemia not treated with a statin.59,67,68 The primary efficacy endpoint is a greater mean percentage LDL-C reduction in alirocumab-treated patients from baseline to 24 weeks (vs statin therapy)59,67,68
ODYSSEY ALTERNATIVE assessed the efficacy and safety of alirocumab compared with ezetimibe in statin-intolerant patients at moderate to very-high ASCVD risk.59 Intolerance was defined as inability to take at least two different statins because of muscle-related adverse effects, one of which must have been administered at the lowest-approved starting dose. Patients first received single-blind subcutaneous or oral placebo for 4 weeks and were withdrawn if they developed muscle-related adverse effects during placebo treatment.69 Continuing patients were randomized (2:2:1 ratio) to alirocumab, 75 mg, every 2 weeks, ezetimibe, 10 mg/d, or atorvastatin, 20 mg/d (statin rechallenge) for 24 weeks. The alirocumab dose was increased to 150 mg every 2 weeks at week 12 if the week 8 LDL-C level was ≤ 70 g/dL in the very high-CV risk patient population or ≥ 100 mg/dL in the medium and high-risk cohort.59,69 The primary endpoint was percentage change in LDL-C from baseline to week 24 by intent- to-treat analysis.59,69 Key findings included (1) a greater percentage reduction from baseline in LDL-C at 24 weeks with alirocumab ver sus ezetimibe (45% vs 14.6%, respectively)69; (2) LDL-C treatment target goal of LDL-C < 100 mg/dL was observed in 61% of ali rocumab- versus 10% of ezetimibe - treated patients (P < .001); and (3) lipoprotein(a) [Lp(a)] was reduced by 25.9% with alirocumab versus 7.3% with ezetimibe (P < .001)7° The rate of study drug discontinuation due to adverse events was lower with alirocumab than with ezetimibe.
The ODYSSEY OPTIONS I and II are randomized, double-blind, active-controlled studies (duration, 24 weeks). The ODYSSEY OPTIONS I trial is evaluating LDL-C level reduction by alirocumab versus ezetimibe when administered as add-on therapy to atorvastatin compared with either doubling the atorvastatin dose or switching from atorvastatin to rosuvastatin.59,71 ODYSSEY OPTIONS II is evaluating LDL-C reduction by alirocumab versus ezetimibe when administered as add-on therapy to rosuvastatin compared with doubling the rosuvastatin dose.59,72 The primary efficacy endpoint was the mean percentage LDL-C reduction in alirocumab- versus statin-treated patients from baseline to week 24. ODYSSEY OPTIONS I showed that the addition of alirocumab as addon to atorvastatin, 20 mg or 40 mg, or to rosuvastatin, 10 mg, produced significantly greater LDL-C reductions versus the addition of ezetimibe, doubling of the statin dose, or switch to rosuvastatin. There was no significant difference observed between adding alirocumab to an entry dose of rosuvastatin of 20 mg or doubling of the rosuvastatin dose.73
The ODYSSEY OUTCOMES trial is evaluating the effect of alirocumab on ASCVD event incidence in patients who have experienced a recent ACS event and are receiving medical and dietary management for dyslipidemia.59,74 The primary endpoint is major adverse CV events (a composite of death, nonfatal MI, ischemic stroke, and unstable angina requiring hospitalization) and the goal is to demonstrate significant reduction in CV outcomes with use of alirocumab.74
ODYSSEY OLE is an open-label study assessing the long-term safety of alirocumab when added to other LLT in patients with HeFH.75 It will also evaluate the long-term efficacy of alirocumab on multiple lipid parameters, as reflected by percentage change from baseline.
Evolocumab Clinical Trials
The key phase III clinical trials for evolocumab are components of the Program to Reduce LDL-C and Cardiovascular Outcomes Following Inhibition of PCSK9 In Different Populations (PROFICIO) to assess LDL-C reduction and ASCVD outcomes. The component trials include the following trials: Goal Achievement After Utilizing an Anti-PCSK9 Antibody in Statin Intolerant Subjects (GAUSS)-2; Reduction of LDL-C With PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD)-2; Monoclonal Antibody Against PCSK9 to Reduce Elevated LDL-C in Patients Currently Not Receiving Drug Therapy for Easing Lipid Levels (MENDEL)-2; LDL-C Assessment With PCSK9 Monoclonal Antibody Inhibition Combined With Statin Therapy (LAPLACE)-2; Durable Effect of PCSK9 Antibody Compared with Placebo (DESCARTES); Trial Evaluating PCSK9 Antibody in Subjects With LDL Receptor Abnormalities (TESLA)-2; Open Label Study of Long Term Evaluation Against LDL-C (OSLER)-2; Further Cardiovascular Outcomes Research With PCSK9 Inhibition In Subjects With Elevated Risk (FOURIER); and Global Assessment of Plaque Regression With a PCSK9 Antibody as Measured by Intravascular Ultrasound (GLAGOV). The FOURIER and GLAGOV trials are ongoing.
The GAUSS-2 trial assessed hyperlipidemic patients who were statin intolerant.76,77 Evolocumab (140 mg every 2 weeks or 420 mg monthly) reduced LDL-C from baseline by 53% and 5 6% when compared with ezetimibe (37%-39% reduction from baseline respectively; P < .001). Muscle-based adverse events occurred in 12% of evolocumab-treated patients versus 23% of ezetimibe-treated patients. Treatment-emergent adverse events and laboratory abnormalities were comparable across treatment groups.77
The RUTHERFORD-2 trial assessed and evaluated safety, tolerability, and efficacy of evolocumab dosing (140 mg every 2 weeks or 420 mg monthly) in HeFH patients.78 Key results showed that, compared with placebo, evolocumab at both dosing schedules showed significant reduction in mean LDL-C at week 12 (every 2 weeks, 59.2%; monthly, 61.3%; both P < .0001). (Figure 9).78 Evolocumab was well tolerated, with similar rates of adverse events as placebo.
The MENDEL-2 trial assessed stand-alone evolocumab treatment in patients with hyperlipid-emia versus ezetimibe or placebo.79 Evolocumab dosage regimens rapidly and markedly lowered LDL-C over 12 weeks compared with either placebo or ezetimibe (Figure 10).79 The overall incidence of treatment-emergent adverse effects was comparable across treatment groups.
The LAPLACE-2 trial assessed adding evolocumab (140 mg every 2 weeks or 420 mg monthly) to medium- or high-intensity statin therapy versus ezetimibe for patients with hyperlipidemia.79 In patients with hypercholesterolemia on background statin therapy, evolocumab every 2 weeks reduced LDL-C concentration between 66% and 75% compared with placebo at 10 to 12 weeks of treatment.80 Evolocumab administered monthly reduced LDL-C concentration between 63% and 75% versus placebo,80 whereas ezetimibe reduced LDL-C by 20% to 25% compared with placebo.80
The DESCARTES trial evaluated the safety and efficacy of evolocumab 420 mg or placebo administered to subjects with fasting LDL-C ≥ 75 mg/dL and a range of CV risk when added to one of four different background LLT groups.81 Evolocumab reduced LDL-C from baseline by 57% (Figure 11)81 and there was a 90% reduction in PCSK9 levels after 1 week on evolocumab. The adverse event profile was similar between the placebo (74.2%) and evolocumab treatment groups (74.8%).81
The TESLA-2 study assessed the safety, tolerability, and efficacy of evolocumab plus a statin in patients with homozygous familial hypercholesterolemia compared with those on statin therapy alone (plus placebo).81 Compared with placebo, evolocumab reduced LDL-C by 30.9% at 12 weeks81 and no serious adverse side effects were noted.82
The OSLER-2 trial is an ongoing study composed of patients from phase III trials and is assessing long-term safety and efficacy of evolocumab in an open-label study.83,84 Evolocumab reduced LDL-C by 52.1%.84 Patients who discontinued evolocumab had LDL-C levels return to near baseline. Adverse and serious adverse events were observed in 81.4% and 7.1%, respectively, of evolocumab-treated patients, versus 73.1% and 6.3%, respectively, of individuals in the standard of care (SOC) group.84 The five most common adverse effects in the evolocumab versus SOC treatment groups were nasopharyngitis (12.2% vs 9.8%), upper respiratory tract infections (7.7% vs 7.6%), influenza (7.1% vs 5.2%), arthralgia (6.9% vs 4.3%), and back pain (6.5% vs 5.4%).84
Two phase III clinical trials are ongoing. The FOURIER trial will assess how evolocumab plus statin therapy compares with statin therapy alone for treating 22,500 high-risk patients with hyperlipidemia.85 The GLAGOV trial will determine the effect of evolocumab on plaque atheroma volume in patients with CAD undergoing cardiac catheterization.86
Bococizumab
Bococizumab is being assessed in phase III clinical trials: the Evaluation of RN316 in Reducing the Occurrence of Major Cardiovascular Events in High Risk Subjects (SPIRE) I and II. The SPIRE I study will assess whether lowering LDL-C to levels well below current guideline-recommended targets will lead to further reduction in CV events in a high-risk patient population with baseline LDL-C levels ranging from 70 to 100 mg/dL.87 The SPIRE II study will evaluate the efficacy and safety of bococizumab in a range of high-risk patients who have not achieved LDL-C levels < 100 mg/dL despite the use of high-dose statins or who are partially or completely statin intolerant.88
Potential Implications of PCSK9 Inhibition and Reductions in Lp(a)
Among nontraditional risk factors for CV disease, Lp(a) merits special mention. As a compound with multiple isoforms, as well as chemical moieties structurally resembling both LDL-C and plasminogen, it is thought to play an important role in atherogenesis and throm-bogenesis via intimal accumulation, activation, and recruitment of macrophages and smooth muscle cells, and inhibition of fibrinolysis.89,90 In an older study by Dangas and colleagues,89 Lp(a) was found to be ubiquitous in atherectomy specimens from patients presenting with ACS, and, together with tissue macrophages, it was found in higher concentrations in those patients with unstable syndromes. This may explain the repeated demonstration of an association between serum Lp(a) levels and CV events.91-94 Notable among these data was the finding by Albers and associates92 of the predictive value of both baseline and on-therapy serum Lp(a) levels for CV events, even in a cohort of dyslipidemic patients treated with statins and niacin versus placebo with or without ezetimibe to a serum LDL-C level of 60 to 80 mg/dL. This finding mirrors the results of a post-hoc analysis by Khera and colleagues95 of data from the Justification for Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) trial database. These investigators found a consistent association between on-treatment serum Lp(a) levels and incident CVD. Both studies suggest that Lp(a) may be a marker for a residual risk for CV events even in patients on statin therapy and at LDL-C targets. The consistent and robust reductions of Lp(a) observed with PCSK9 inhibitors may represent an added benefit above and beyond the concurrent reductions in LDL-C and non-HDL-C levels.
Is There a Cholesterol Level That Is Too Low?
There have been reports of healthy individuals documented with LDL-C levels as low as 15 mg/dL due to genetic mutations.96 PCSK9 inhibitor studies are ongoing, and will determine if LDL-C lowering beyond what is observed with statin therapy provides incremental and safe CV protection. The LDL-C levels recommended in clinical trials may be as low as 25 to 50 mg/dL.96 In data from ODYSSEY Long Term, in 562 patients who had two consecutive LDL-C measures < 25 mg/dL, there was no signal of any increase in treatment-emergent adverse events of special interest including neurologic and CV events or neurocognitive disorders over the course of this 18-month study.97
Hypercholesterolemia Management and 2013 ACC/AHA Cholesterol Guidelines
The 2013 ACC/AHA guidelines recommend measuring LDL-C percentage change from baseline over LDL-C numerical target values based on CV risk.29 The guidelines show several groups in whom the benefits of moderate- to high-intensity statin monotherapy outweigh the risks29: individuals with clinical ASCVD, individuals with baseline LDL-C ≥ 190 mg/dL, individuals age 40 to 75 years with type I or II diabetes and LDL-C between 70 and 189 mg/dL without clinical ASCVD, and individuals age 40 to 75 years without clinical ASCVD or type I or II diabetes and LDL-C between 70 and 189 mg/dL and an estimated 10-year ASCVD risk calculation of ≥ 7.5%. The guidelines also identify several groups in which statin benefits are not supported by clinical data (but can be considered according to clinical judgment)29: patients age < 40 years, patients age >75 years, hemodialysis patients, and patients with congestive heart failure. The guidelines suggested nonstatin therapies be considered for the following groups29: patients who do not respond as expected to statins, patients who cannot tolerate recommended statin dose intensity, and patients who have statin intolerance.
Use of PCSK9 Inhibitors in Relation to 2013 ACC/AHA Guidelines
The 2013 ACC/AHA guidelines downplayed, to some extent, the potential nonstatin value for achieving desired LDL-C or non-HDL-C treatment goals because RCT evidence has not yet supported their use.29 However, the guidelines also identified high-priority research needs, including alternative ASCVD risk-reduction strategies for patients intolerant of or resistant to statin therapy. Since publication of the guidelines, PCSK9 inhibitors have shown efficacy in lowering LDL-C levels while demonstrating their safety in use in numerous clinical trials. Thus, PCSK9 inhibitors may provide an effective treatment for patients who are statin intolerant or who do not reach therapeutic goals due to genetic abnormalities such as HeFH.
Conclusions
To reduce CV risk, LDL-C reduction remains the primary target for lipid modification. Many patients, including those with HeFH, are not able to reach LDL-C treatment goals with statins due to intolerance or resistance. PCSK9 inhibitors increase the functional half-life of the LDL-R and lead to increased removal of LDL-C from the circulation. PCSK9 inhibitors are effective either as monotherapy or as an adjunct to other LLTs and reduce LDL-C levels by 50% to 70%. Furthermore, PCSK9 mAbs appear to be well tolerated and safe. Signals are appearing in clinical trials that suggest a substantial incremental reduction of CV events with PCSK9 inhibition above and beyond that related to statins. Large RCTs with CV events as the primary endpoint are in progress (ODYSSEY OUTCOMES, FOURIER, and SPIRE) and should provide us with a wealth of information on their impact on CV events. ![]()
Dr. Kereiakes, Dr. Contreras, and Dr. Desai have no real or apparent conflicts of interest. Dr. Lepor has received grant/research support from sanofiaventis (Bridgewater, NJ), Regeneron (Tarrytown, NY), and Amgen (Thousand Oaks, CA), and honoraria from sanofiaventis and Regeneron.
References
- Yusuf S, Hawken S, Ounpuu S, et al; INTERHEART Study Investigators. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364:937-952.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics--2014 update: a report from the American Heart Association. Circulation. 2014;129:e28-e292.
- Kannel WB, Castelli WP, Gordon T, McNamara PM. Serum cholesterol, lipoproteins, and the risk of coronary heart disease. The Framingham study. Ann Intern Med. 1971;74:1-12.
- Neaton JD, Wentworth D. Serum cholesterol, blood pressure, cigarette smoking, and death from coronary heart disease. Overall findings and differences by age for 316,099 white men. Multiple Risk Factor Intervention Trial Research Group. Arch Intern Med. 1992;152:56-64.
- Newman WP 3rd, Freedman DS, Voors AW, et al. Relation of serum lipoprotein levels and systolic blood pressure to early atherosclerosis. The Bogalusa Heart Study. N Engl J Med. 1986;314:138-144.
- Clofibrate and niacin in coronary heart disease. JAMA. 1975;231:360-381.
- Lipid Research Clinics Coronary Primary Prevention Trial results. I. Reduction in incidence of coronary heart disease. JAMA. 1984;251:351-364.
- Buchwald H, Varco RL, Matts JP, et al. Effect of partial ileal bypass surgery on mortality and morbidity from coronary heart disease in patients with hypercholesterolemia. Report of the Program on the Surgical Control of the Hyperlipidemias (POSCH). N Engl J Med. 1990;323:946-955.
- O’Keefe JH Jr, Cordain L, Harris WH, et al. Optimal low-density lipoprotein is 50 to 70 mg/dl: lower is better and physiologically normal. J Am Coll Cardiol. 2004;43:2142-2146.
- Wiviott SD, Cannon CP, Morrow DA, et al; PROVE IT-TIMI 22 Investigators. Can low-density lipoprotein be too low? The safety and efficacy of achieving very low low-density lipoprotein with intensive statin therapy: a PROVE IT-TIMI 22 substudy. J Am Coll Cardiol. 2005;46:1411-1416.
- Assmann G, Gotto AM Jr. HDL cholesterol and protective factors in atherosclerosis. Circulation. 2004;109(suppl):III8-III14.
- A Study of AMR101 to Evaluate Its Ability to Reduce Cardiovascular Events in High Risk Patients With Hypertriglyceridemia and on Statin. The Primary Objective is to Evaluate the Effect of 4 g/Day AMR101 for Preventing the Occurrence of a First Major Cardiovascular Event. (REDUCE-IT). ClinicalTrials.gov Web site. http://clinicaltrials.gov/show/NCT01492361. Accessed December 1, 2014.
- Nordestgaard BG, Chapman MJ, Humphries SE, et al; European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478-3490.
- Pijlman AH, Huijgen R, Verhagen SN, et al. Evaluation of cholesterol lowering treatment of patients with familial hypercholesterolemia: a large cross-sectional study in The Netherlands. Atherosclerosis. 2010;209:189-194.
- Goldstein J, Schrott HG, Hazzard WR, et al. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest. 1973;52:1544-1568.
- Marks D, Thorogood M, Neil HA, Humphries SE. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis. 2003;168:1-14.
- Jarvik GP, Brunzell JD, Motulsky AG. Frequent detection of familial hypercholesterolemia mutations in familial combined hyperlipidemia. J Am Coll Cardiol. 2008;52:1554-1556.
- National Institute for Health and Care Excellence. Identification and management of familial hypercholesterolaemia. NICE Clinical Guideline CG71; August 2008. National Institute for Health and Care Excellence Web site. http://www.nice.org.uk/guidance/cg71. Accessed December 2, 2014.
- Civeira F, Castillo S, Alonso R, et al; Spanish Familial Hypercholesterolemia Group. Tendon xanthomas in familial hypercholesterolemia are associated with cardiovascular risk independently of the low-density lipoprotein receptor gene mutation. Arterioscler Thromb Vasc Biol. 2005;25:1960-1965.
- Civeira F, Jarauta E, Cenarro A, et al. Frequency of low-density lipoprotein receptor gene mutations in patients with a clinical diagnosis of familial combined hyperlipidemia in a clinical setting. J Am Coll Cardiol. 2008;52:1546-1553.
- Cohen JD, Brinton EA, Ito MK, Jacobson TA. Understanding Statin Use in America and Gaps in Patient Education (USAGE): an internet-based survey of 10,138 current and former statin users. J Clin Lipidol. 2012;6:208-215.
- Rodriguez F, Olufade T, Heithoff K, et al. Frequency of high-risk patients not receiving high potency statin (from a large U.S. managed care database). Am J Cardiol. 2015;115:190-195.
- Baigent C, Landray MJ, Reith C, et al; SHARP Investigators. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomized placebo-controlled trial. Lancet. 2011;377: 2181-2192.
- Postmus I, Trompet S, Deshmukh HA, et al. Pharmacogenetic meta-analysis of genome-wide association studies of LDL cholesterol response to statins. Nat Commun. 2014;5:5068.
- Elshazly MB, Martin SS, Blaha MJ, et al. Non-high density lipoprotein cholesterol, guideline targets, and population percentiles for secondary prevention in 1.3 million adults: the VLDL-2 study (very large database of lipids). J Am Coll Cardiol. 2013;62:1960-1965.
- Mora S, Wenger NK, Demicco DA, et al. Determinants of residual risk in secondary prevention patients treated with high- versus low-dose statin therapy: the Treating to New Targets (TNT) study. Circulation. 2012;125:1979-1987.
- Jia L, Betters JL, Yu L. Niemann-pick C1-like 1 protein in intestinal and hepatic cholesterol transport. Annu Rev Physiol. 2011;73:239-259.
- Cannon CP. IMPROVE-IT Trial: a comparison of ezetimibe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes after acute coronary syndromes. Presented at: American Heart Association Scientific Sessions 2014; November 15-19, 2014; Chicago, IL.
- Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/ AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/ American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 Pt B): 2889-2934.
- Ray KK, Kastelein JJ, Boekholdt SM, et al. The ACC/AHA 2013 guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular disease risk in adults: the good the bad and the uncertain: a comparison with ESC/EAS guidelines for the management of dyslipidaemias 2011. Eur Heart J. 2014;35:960-968.
- Lambert G, Sjouke B, Choque B, et al. The PCSK9 decade. J Lipid Res. 2012;53:2515-2524.
- Lagace TA, Curtis DE, Garuti R, et al. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest. 2006;116:2995-3005.
- Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154-156.
- Rashid S, Curtis DE, Garuti R, et al. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking PCSK9. Proc Natl Acad Sci U S A. 2005; 5374-5379.
- Cohen J, Pertsemlidis A, Kotowski IK, et al. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37:161-165.
- Cohen J, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264-1272.
- Horton JD, Shah NA, Warrington JA, et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci USA. 2003;100: 12027-12032.
- Attie AD. The mystery of PCSK9. Arterioscl Thromb Vasc Biol. 2004;24:1337-1339.
- Davidson MH. Emerging therapies for lowering LDL-C: focus on PCSK9 monoclonal antibodies. Medscape Web site. http://www.medscape.org/viewarticle/763848_transcript. Accessed December 1, 2014.
- Qian YW, Schmidt RJ, Zhang Y, et al. Secreted PCSK9 downregulates low density lipoprotein receptor through receptor-mediated endocytosis. J Lipid Res. 2007;48:1488-1498.
- Seidah NG, Mayer G, Zaid A, et al. The activation and physiological functions of the proprotein convertases. Int J Biochem Cell Biol. 2008;40:1111-1125.
- Maxwell KN, Soccio RE, Duncan EM, et al. Novel putative SREBP and LXR target genes identified by microarray analysis in liver of cholesterol-fed mice. J Lipid Res. 2003;44:2109-2119.
- Sato R. Sterol metabolism and SREBP activation. Arch Biochem Biophys. 2010;501:177-181.
- Maxwell KN, Breslow JL. Antibodies to PCSK9: a superior way to lower LDL cholesterol? Circ Res. 2012;111:274-277.
- Lambert G, Charlton F, Rye KA, Piper DE. Molecular basis of PCSK9 function. Atherosclerosis. 2009;203:1-7.
- Lipovsek D. Adnectins: engineered target-binding protein therapeutics. Protein Eng Des Sel. 2011;24:3-9.
- Fitzgerald K, Frank-Kamenetsky M, Shulga-Morskaya S, et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet. 2014;383:60-68.
- Hunt SC, Hopkins PN, Bulka K, et al. Genetic localization to chromosome 1p32 of the third locus for familial hypercholesterolemia in a Utah kindred. Arterioscler Thromb Vasc Biol. 2000;20:1089-1093.
- Timms KM, Wagner S, Samuels ME, et al. A mutation in PCSK9 causing autosomal-dominant hypercholesterolemia in a Utah pedigree. Hum Genet. 2004;114:349-353.
- Benn M, Nordestgaard BG, Grande P, et al. PCSK9 R46L, low-density lipoprotein cholesterol levels, and risk of ischemic heart disease: 3 independent studies and meta-analyses. J Am Coll Cardiol. 2010;55: 2833-2842.
- Catapano AL, Papadopoulos N. The safety of therapeutic monoclonal antibodies: implications for cardiovascular disease and targeting the PCSK9 pathway. Atherosclerosis. 2013;228:18-28.
- Dias CS, Shaywitz AJ, Wasserman SM, et al. Effects of AMG 145 on low-density lipoprotein cholesterol levels: results from 2 randomized, double-blind, placebo- controlled, ascending-dose phase 1 studies in healthy volunteers and hypercholesterolemic subjects on statins. J Am Coll Cardiol. 2012;60:1888-1898.
- Stein EA, Mellis S, Yancopoulos GD, et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366:1108-1118.
- Gumbiner B. Effects of 12 weeks of treatment with RN316 (PF-04950615), a humanized Ig2a monoclonal binding protein convertase subtilisin kexin type 9, in hypercholesterolemic subjects on high and maximal dose statins. Presented at: American Heart Association Scientific Sessions; November 3-7, 2012; Los Angeles, CA.
- McKenney JM, Koren MJ, Kereiakes DJ, et al. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am Coll Cardiol. 2012;59: 2344-2353.
- Roth EM, McKenney JM, Hanotin C, et al. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med. 2012;367:1891-1900.
- Stein EA, Gipe D, Bergeron J, et al. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380:29-36.
- Ginsberg HN, Rader DJ, Raal FJ, et al. ODYSSEY HIGH FH: efficacy and safety of alirocumab in patients with severe heterozygous familial hypercholesterolemia. Presented at: American Heart Association Scientific Sessions 2014; November 15-19, 2014; Chicago, IL.
- Roth EM, Diller P. Alirocumab for hyperlipidemia: physiology of PCSK9 inhibition, pharmacodynamics and Phase I and II clinical trial results of a PCSK9 monoclonal antibody. Fut Cardiol. 2014;10: 183-199.
- Efficacy and Safety of Alirocumab (SAR236553/ REGN727) Versus Placebo on Top of Lipid-Modifying Therapy in Patients With Heterozygous Familial Hypercholesterolemia Not Adequately Controlled With Their Lipid-Modifying Therapy. (ODYSSEY FH I). ClinicalTrials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT01623115?term=Odyssey+FH+I&">http://clinicaltrials.gov/ct2/show/NCT01623115?term=Odyssey+FH+I&rank=1" target="_blank">http://clinicaltrials.gov/ct2/show/NCT01623115?term=Odyssey+FH+I&rank=1. Accessed December 1, 2014.
- Study of Alirocumab (REGN727/SAR236553) in Patients With heFH (Heterozygous Familial Hypercholesterolemia) Who Are Not Adequately Controlled With Their LMT (Lipid-Modifying Therapy). (Odyssey FH II). ClinicalTrials.gov Web site. http://clinicaltrials.gov/show/NCT01709500. Accessed December 1, 2014.
- Roth EM, Taskinen MR, Ginsberg HN, et al. Monotherapy with the PCSK9 inhibitor alirocumab versus ezetimibe in patients with hypercholesterolemia: results of a 24 week, double-blind, randomized Phase 3 trial. Int J Cardiol. 2014;176:55-61.
- Colhoun HM, Robinson JG, Farnier M, et al. Efficacy and safety of alirocumab, a fully human PCSK9 monoclonal antibody, in high cardiovascular risk patients with poorly controlled hypercholesterolemia on maximally tolerated doses of statins: rationale and design of the ODYSSEY Combo I and II trials. BMC Cardiovasc Disord. 2014;14:121.
- Efficacy and Safety of Alirocumab (SAR236553/ REGN727) Versus Ezetimibe on Top of Statin in High Cardiovascular Risk Patients With Hypercholesterolemia. (ODYSSEY Combo II). ClinicalTrials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT01644188?term=Odyssey+COMBO+II&">http://clinicaltrials.gov/ct2/show/NCT01644188?term=Odyssey+COMBO+II&rank=1" target="_blank">http://clinicaltrials.gov/ct2/show/NCT01644188?term=Odyssey+COMBO+II&rank=1. Accessed December 1, 2014.
- Kereiakes DJ, Robinson JG, Cannon CP, et al. Efficacy and safety of alirocumab in high cardiovascular risk patients with suboptimally controlled hypercholesterolemia on maximally tolerated doses of statins: the ODYSSEY COMBO I study. Presented at: American Heart Association Scientific Sessions 2014; November 15-19, 2014; Chicago, IL.
- Long-term Safety and Tolerability of Alirocumab SAR236553 (REGN727) Versus Placebo on Top of Lipid-Modifying Therapy in High Cardiovascular Risk Patients With Hypercholesterolemia. (ODYSSEY Long Term). ClinicalTrials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT01507831?term=ODYSSEY+LONG+TERM&">http://clinicaltrials.gov/ct2/show/NCT01507831?term=ODYSSEY+LONG+TERM&rank=1" target="_blank">http://clinicaltrials.gov/ct2/show/NCT01507831?term=ODYSSEY+LONG+TERM&rank=1. Accessed December 1, 2014.
- Study to Evaluate the Efficacy and Safety of an Every Four Weeks Treatment Regimen of Alirocumab (REGN727/ SAR236553) in Patients With Primary Hypercholesterolemia. (ODYSSEY CHOICE I). ClinicalTrials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT01926782?term=ODYSSEY+CHOICE&">http://clinicaltrials.gov/ct2/show/NCT01926782?term=ODYSSEY+CHOICE&rank=2" target="_blank">http://clinicaltrials.gov/ct2/show/NCT01926782?term=ODYSSEY+CHOICE&rank=2. Accessed December 1, 2014.
- Phase III Study To Evaluate Alirocumab in Patients With Hypercholesterolemia Not Treated With a Statin. (ODYSSEY CHOICE II). ClinicalTrials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT02023879?term=CHOICE+II&">http://clinicaltrials.gov/ct2/show/NCT02023879?term=CHOICE+II&rank=2" target="_blank">http://clinicaltrials.gov/ct2/show/NCT02023879?term=CHOICE+II&rank=2. Accessed December 1, 2014.
- Study of Alirocumab (REGN727/ SAR236553) in Patients With Primary Hypercholesterolemia and Moderate, High, or Very High Cardiovascular (CV) Risk, Who Are Intolerant to Statins. (Odyssey Alternative). ClinicalTrials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT01709513?term=ODYSSEY+Alternative&">http://clinicaltrials.gov/ct2/show/NCT01709513?term=ODYSSEY+Alternative&rank=1" target="_blank">http://clinicaltrials.gov/ct2/show/NCT01709513?term=ODYSSEY+Alternative&rank=1. Accessed December 1, 2014.
- Moriarty P, Thompson PD, Cannon CP, et al. ODYSSEY ALTERNATIVE: efficacy and safety of the proprotein convertase subtilisin/kexin type 9 monoclonal antibody, alirocumab, versus ezetimibe, in patients with statin intolerance as defined by a placebo run-in and statin rechallenge arm. Presented at: American Heart Association Scientific Sessions 2014; November 15-19, 2014; Chicago, IL.
- Study of the Efficacy and Safety of REGN727 (SAR236553) in Combination With Other Lipidmodifying Treatments (LMT). ClinicalTrials.gov Web site. http://clinicaltrials.gov/show/NCT01730040. Accessed December 1, 2014.
- Study of Alirocumab (REGN727/SAR236553) addedon to Rosuvastatin Versus Other Lipid Modifying Treatments (LMT). ClinicalTrials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT01730053?term=REGN727&">http://clinicaltrials.gov/ct2/show/NCT01730053?term=REGN727&phase=2&rank=8" target="_blank">http://clinicaltrials.gov/ct2/show/NCT01730053?term=REGN727&phase=2&rank=8. Accessed December 1, 2014.
- Bays H, Farnier M, Gaudet D, et al. Efficacy and safety of combining alirocumab with atorvastatin or rosuvastatin versus adding ezetimibe, doubling statin dose or switching statin therapy in high cardiovascular risk patients: Odyssey Options I and II. Presented at: American Heart Association Scientific Sessions 2014; November 15-19, 2014; Chicago, IL.
- ODYSSEY Outcomes: Evaluation of Cardiovascular Outcomes After an Acute Coronary Syndrome During Treatment With Alirocumab SAR236553. (REGN727). ClinicalTrials.gov Web site. http://www.clinicaltrials.gov/ct2/show/NCT01663402?term=ODYSSEY+OUTCOMES&">http://www.clinicaltrials.gov/ct2/show/NCT01663402?term=ODYSSEY+OUTCOMES&rank=1" target="_blank">http://www.clinicaltrials.gov/ct2/show/NCT01663402?term=ODYSSEY+OUTCOMES&rank=1. Accessed December 1, 2014.
- Open Label Study of Long Term Safety Evaluation of Alirocumab. (ODYSSEY OLE). ClinicalTrials.gov Web site. http://clinicaltrials.gov/show/NCT01954394. Accessed December 1, 2014.
- Cho L, Rocco M, Colquhoun D, et al. Design and rationale of the GAUSS-2 study trial: a double-blind, ezetimibe-controlled phase 3 study of the efficacy and tolerability of evolocumab (AMG 145) in subjects with hypercholesterolemia who are intolerant of statin therapy. Clin Cardiol. 2014;37:131-139.
- Stroes E, Colquhoun D, Sullivan D, et al; GAUSS-2 Investigators. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2541-2548.
- Raal FJ, Stein EA, DuFour R, et al; RUTHERFORD-2 Investigators. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double- blind, placebo-controlled trial [published online ahead of print October 1, 2014]. Lancet. doi: 10.1016/ S0140-6736(14)61399-4.
- Koren MJ, Lundqvist P, Bolognese M, et al; MENDEL- 2 Investigators. Anti-PCSK9 monotherapy for hypercholesterolemia: the MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2531-2540.
- Robinson JG, Nedergaard BS, Rogers WJ, et al; LAPLACE- 2 Investigators. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA. 2014;311:1870-1882.
- Blom DJ, Hala T, Bolognese M, et al; DESCARTES Investigators. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med. 2014;370:1809-1819.
- Raal FJ, Honarpour N, Blom DJ, et al; for the TESLA Investigators. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial [published ahead of print October 1, 2014]. Lancet.. doi: 10.1016/S0140-6736(14) 61374-X.
- Koren MJ, Giugliano RP, Raal FJ, et al; OSLER Investigators. Efficacy and safety of longer-term administration of evolocumab (AMG 145) in patients with hypercholesterolemia: 52-week results from the Open-Label Study of Long-Term Evaluation Against LDL-C (OSLER) randomized trial. Circulation. 2014;129:234-243.
- Open Label Study of Long Term Evaluation Against LDL-C Trial-2. (OSLER-2). ClinicalTrials.gov Web site. http://clinicaltrials.gov/show/NCT01854918. Accessed December 1, 2014.
- Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects With Elevated Risk. (FOURIER). ClinicalTrials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT01764633?term=NCT01764633&">http://clinicaltrials.gov/ct2/show/NCT01764633?term=NCT01764633&rank=1" target="_blank">http://clinicaltrials.gov/ct2/show/NCT01764633?term=NCT01764633&rank=1. Accessed December 1, 2014.
- GLobal Assessment of Plaque reGression With a PCSK9 antibOdy as Measured by intraVascular Ultrasound. (GLAGOV). ClinicalTrials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT01813422?term=NCT01813422&">http://clinicaltrials.gov/ct2/show/NCT01813422?term=NCT01813422&rank=1" target="_blank">http://clinicaltrials.gov/ct2/show/NCT01813422?term=NCT01813422&rank=1. Accessed December 1, 2014.
- The Evaluation of Bococizumab (PF-04950615) in Reducing the Occurrence of Major Cardiovascular Events in High Risk Subjects. (SPIRE-1). ClinicalTrials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT01975376?term=SPIRE-1&">http://clinicaltrials.gov/ct2/show/NCT01975376?term=SPIRE-1&rank=1" target="_blank">http://clinicaltrials.gov/ct2/show/NCT01975376?term=SPIRE-1&rank=1. Accessed December 1, 2014.
- The Evaluation of Bococizumab (PF-04950615) in Reducing the Occurrence of Major Cardiovascular Events in High Risk Subjects. (SPIRE-2). ClinicalTrials.gov Web site. http://clinicaltrials.gov/ct2/show/NCT01975389?term=SPIRE-2&">http://clinicaltrials.gov/ct2/show/NCT01975389?term=SPIRE-2&rank=1" target="_blank">http://clinicaltrials.gov/ct2/show/NCT01975389?term=SPIRE-2&rank=1. Accessed December 1, 2014.
- Dangas G, Mehran R, Harpel PC, et al. Lipoprotein(a) and inflammation in human coronary atheroma: association with the severity of clinical presentation. J Am Coll Cardiol. 1998;32:2035-2042.
- Tsimikas S, Hall JL. Lipoprotein(a) as a potential causal genetic risk factor of cardiovascular disease: a rationale for increased efforts to understand its pathophysiology and develop targeted therapies. J Am Coll Cardiol. 2012;60:716-721.
- O’Donoghue ML, Morrow DA, Tsimikas S, et al. Lipoprotein(a) for risk assessment in patients with established coronary artery disease. J Am Coll Cardiol. 2014;63:520-527.
- Albers JJ, Slee A, O’Brien KD, et al. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J Am Coll Cardiol. 2013;62:1575-1579.
- Alonso R, Andres E, Mata N, et al; SAFEHEART Investigators. Lipoprotein(a) levels in familial hypercholesterolemia: an important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J Am Coll Cardiol. 2014;63: 1982-1989.
- Willeit P, Kiechl S, Kronenberg F, et al. Discrimination and net reclassification of cardiovascular risk with lipoprotein(a): prospective 15-year outcomes in the Bruneck Study. J Am Coll Cardiol. 2014;64:851-860.
- Khera AV, Everett BM, Caulfield MP, et al. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). Circulation. 2014;129:635-642.
- Kolata G. Rare mutation ignites race for cholesterol drug. New York Times. July 10, 2013:A1. http://www.nytimes.com/2013/07/10/health/rare-mutationprompts-race-for-cholesterol-drug.html?pagewanted=3&">http://www.nytimes.com/2013/07/10/health/rare-mutationprompts-race-for-cholesterol-drug.html?pagewanted=3&ref=ginakolata&pagewanted=all" target="_blank">http://www.nytimes.com/2013/07/10/health/rare-mutationprompts-race-for-cholesterol-drug.html?pagewanted=3&ref=ginakolata&pagewanted=all. Accessed December 1, 2014.
- Robinson JG, Farnier M, Krempf M, et al. Long-term safety, tolerability and efficacy of alirocumab in high cardiovascular risk patients: Odyssey Long Term. Presented at: American Heart Association Scientific Sessions 2014; November 15-19, 2014; Chicago, IL.