Original Article
Stability of Reconstituted Telavancin Drug Product in Frozen Intravenous Bags
Zhengtian Gu, PhD*; Anissa Wong, MS†; Elvira Raquinio‡; and Alice Nguyen, BA§
Original Article
Stability of Reconstituted Telavancin Drug Product in Frozen Intravenous Bags
Zhengtian Gu, PhD*; Anissa Wong, MS†; Elvira Raquinio‡; and Alice Nguyen, BA§
Original Article
Stability of Reconstituted Telavancin Drug Product in Frozen Intravenous Bags
Zhengtian Gu, PhD*; Anissa Wong, MS†; Elvira Raquinio‡; and Alice Nguyen, BA§
Abstract
Background and Objective: Intravenous (IV) infusions of telavancin for injection are generally administered in-hospital, but in some circumstances they may be administered in an outpatient environment. In that setting, antibiotics may be premixed and frozen. This study determined the chemical stability of nonpreserved telavancin in various commonly used reconstitution diluents stored in IV bags (polyvinyl chloride [PVC] and PVC-free) at -20°C (-4°F) without light.
Methods: Telavancin (750 mg/vial) was reconstituted with 5% dextrose injection USP (D5W) or 0.9% sodium chloride injection USP (NS) to obtain drug solutions at approximately 15 mg/mL. Infusion solutions of telavancin at diluted concentrations of 0.6 mg/mL and 8.0 mg/mL covering the range utilized in clinical practice were prepared in both PVC and PVC-free IV bags using D5W or NS solutions. The infusion solutions were stored under frozen conditions (-20°C ± 5°C [-4°F ± 41°F]) and the chemical stability was evaluated for up to 32 days. Telavancin concentration, purity, and degradant levels were determined using a stability-indicating high-performance liquid chromatography (HPLC) method.
Results: Telavancin IV infusion solutions in D5W or NS at 0.6 mg/mL and 8 mg/mL and stored at -20°C (-4°F) met the chemical stability criteria when tested on days 0, 7, 14, and 32. The assayed telavancin concentration at each time point was within 97% to 103% of the initial mean assay value. The total degradants quantified by the HPLC stability-indicating method did not show any significant change over the 32-day study period.
Conclusion: Telavancin IV infusion solutions (in D5W or NS) in both PVC and PVC-free IV bags were stable for at least 32 days when stored at -20°C (-4°F) without light. These results provide prolonged frozen stability data further to that previously established for 7 days under refrigerated conditions (2°C-8°C [36°F -46°F]), and for 12 hours at room temperature when diluted into IV bags containing D5W, NS, or lactated Ringer’s solution.
Key Words—frozen IV bag, stability, telavancin for injection
Hosp Pharm—2015;50:609–614
Abstract
Background and Objective: Intravenous (IV) infusions of telavancin for injection are generally administered in-hospital, but in some circumstances they may be administered in an outpatient environment. In that setting, antibiotics may be premixed and frozen. This study determined the chemical stability of nonpreserved telavancin in various commonly used reconstitution diluents stored in IV bags (polyvinyl chloride [PVC] and PVC-free) at -20°C (-4°F) without light.
Methods: Telavancin (750 mg/vial) was reconstituted with 5% dextrose injection USP (D5W) or 0.9% sodium chloride injection USP (NS) to obtain drug solutions at approximately 15 mg/mL. Infusion solutions of telavancin at diluted concentrations of 0.6 mg/mL and 8.0 mg/mL covering the range utilized in clinical practice were prepared in both PVC and PVC-free IV bags using D5W or NS solutions. The infusion solutions were stored under frozen conditions (-20°C ± 5°C [-4°F ± 41°F]) and the chemical stability was evaluated for up to 32 days. Telavancin concentration, purity, and degradant levels were determined using a stability-indicating high-performance liquid chromatography (HPLC) method.
Results: Telavancin IV infusion solutions in D5W or NS at 0.6 mg/mL and 8 mg/mL and stored at -20°C (-4°F) met the chemical stability criteria when tested on days 0, 7, 14, and 32. The assayed telavancin concentration at each time point was within 97% to 103% of the initial mean assay value. The total degradants quantified by the HPLC stability-indicating method did not show any significant change over the 32-day study period.
Conclusion: Telavancin IV infusion solutions (in D5W or NS) in both PVC and PVC-free IV bags were stable for at least 32 days when stored at -20°C (-4°F) without light. These results provide prolonged frozen stability data further to that previously established for 7 days under refrigerated conditions (2°C-8°C [36°F -46°F]), and for 12 hours at room temperature when diluted into IV bags containing D5W, NS, or lactated Ringer’s solution.
Key Words—frozen IV bag, stability, telavancin for injection
Hosp Pharm—2015;50:609–614
Abstract
Background and Objective: Intravenous (IV) infusions of telavancin for injection are generally administered in-hospital, but in some circumstances they may be administered in an outpatient environment. In that setting, antibiotics may be premixed and frozen. This study determined the chemical stability of nonpreserved telavancin in various commonly used reconstitution diluents stored in IV bags (polyvinyl chloride [PVC] and PVC-free) at -20°C (-4°F) without light.
Methods: Telavancin (750 mg/vial) was reconstituted with 5% dextrose injection USP (D5W) or 0.9% sodium chloride injection USP (NS) to obtain drug solutions at approximately 15 mg/mL. Infusion solutions of telavancin at diluted concentrations of 0.6 mg/mL and 8.0 mg/mL covering the range utilized in clinical practice were prepared in both PVC and PVC-free IV bags using D5W or NS solutions. The infusion solutions were stored under frozen conditions (-20°C ± 5°C [-4°F ± 41°F]) and the chemical stability was evaluated for up to 32 days. Telavancin concentration, purity, and degradant levels were determined using a stability-indicating high-performance liquid chromatography (HPLC) method.
Results: Telavancin IV infusion solutions in D5W or NS at 0.6 mg/mL and 8 mg/mL and stored at -20°C (-4°F) met the chemical stability criteria when tested on days 0, 7, 14, and 32. The assayed telavancin concentration at each time point was within 97% to 103% of the initial mean assay value. The total degradants quantified by the HPLC stability-indicating method did not show any significant change over the 32-day study period.
Conclusion: Telavancin IV infusion solutions (in D5W or NS) in both PVC and PVC-free IV bags were stable for at least 32 days when stored at -20°C (-4°F) without light. These results provide prolonged frozen stability data further to that previously established for 7 days under refrigerated conditions (2°C-8°C [36°F -46°F]), and for 12 hours at room temperature when diluted into IV bags containing D5W, NS, or lactated Ringer’s solution.
Key Words—frozen IV bag, stability, telavancin for injection
Hosp Pharm—2015;50:609–614
Hosp Pharm 2015;50(7):609–614
2015 © Thomas Land Publishers, Inc.
doi: 10.1310/hpj5007-609
Telavancin is a lipoglycopeptide antibiotic with bactericidal activity against gram-positive organisms resulting from a dual mechanism of action that inhibits bacterial cell wall synthesis and disrupts membrane barrier function.1 Telavancin for injection is approved in the United States and Canada as a once-daily treatment for adult patients with complicated skin and skin-structure infections (cSSSI) due to gram-positive pathogens including methicillin-resistant Staphylococcus aureus (MRSA)2-4 and in the United States and Europe for the treatment of adult patients with hospital-acquired and ventilator-associated bacterial pneumonia caused by susceptible isolates of S. aureus, when alternative treatments are not suitable.3,5 Telavancin for injection is supplied in single-use vials containing either 250 or 750 mg telavancin as a sterile, lyophilized powder.
The stability of telavancin intravenous (IV) infusion solutions when diluted into IV bags containing 5% dextrose (D5W), 0.9% sodium chloride solution (NS), or lactated Ringer’s solution (LR)6 under refrigerated conditions (2°C-8°C [36°F -46°F]) for 7 days or at room temperature for 12 hours was establishedand approved by the US Food and Drug Administration (FDA) for incorporation into the product label. The purpose of this additional study was to determine the chemical stability of reconstituted telavancin IV infusion solution after storage at frozen conditions (-20°C ± 5°C [-4°F ± 41°F]) for up to 32 days.
METHODS
Materials
Telavancina 750 mg (Lot 2544-90-2029223) vials were used to prepare the samples. D5Wb,c, NSd,e (100 mL), ViaFlex (polyvinyl chloride [PVC]) IV bags and PVC-free IV bags, and sterile syringes (30 cc and 60 cc)f were obtained commercially. Acetonitrile was high-performance liquid chromatography (HPLC) grade or betterg and formic acid was American Chemical Society reagent gradeh. All reagents were used without further purification. Purified water (analytical grade [resistivity of ≥18 MΩ-cm]) was generated in-housei.
Sample Preparation for Chemical Analysis
Samples of telavancin in the drug product vial with butyl rubber stoppers were reconstituted in triplicate with appropriate amounts of the reconstitution diluents (D5W or NS) to yield a telavancin concentration of 15 mg/mL. Vial reconstitution to 15 mg/mL is described in the telavancin label.
Intravenous infusion solutions at target concentrations of 0.6 and 8 mg/mL, which covers the range of concentrations used in clinical practice as specified in the telavancin label, were prepared in triplicate by transferring the required amount of reconstituted telavancin solutions to IV infusion bags of D5W or NS using sterile syringes. Samples reconstituted with D5W and NS were further diluted with D5W and NS, respectively. The IV infusion bag target concentrations were calculated based on theoretical fill volumes of the IV bags; typical overfill volumes were not estimated.
Test Conditions and Drug Attributes Tested
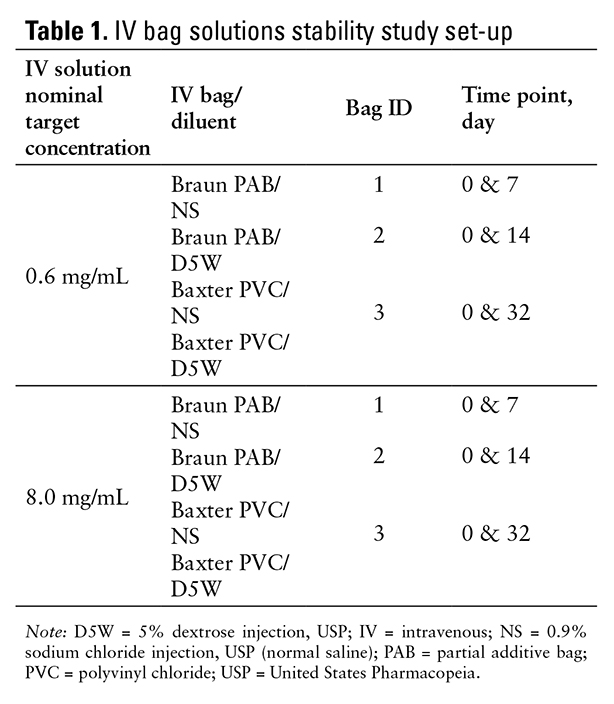
The sample IV bags (uncovered) were stored at -20°C ± 5°C (-4°F ± 41°F) after the initial samples were removed for testing; at the given time point, the IV bags were thawed without any assistance, such as sonication or a hot water bath, until they reached room temperature. The solution was then immediately analyzed. Stability assessments were at the initial time point and either 7, 14, or 32 days for each of the frozen bags to avoid multiple freeze and thaw cycles (refer to Table 1 for the stability study design). The selection of the stability study duration was based on the potential required duration of frozen storage of IV bags at infusion centers or the pharmacies, along with previous stability results of telavancin reconstituted solutions.
The drug attributes tested and the corresponding acceptance criteria were based on the FDA-approved specification: visual appearance of dosage solution (conforms to United States Pharmacopeia [USP]); telavancin concentration by HPLC (90.0%-110.0% of initial assay mean value); level of degradant A by HPLC (≤1.0% w/w); level of degradant B (the primary degradation product of telavancin) by HPLC (≤3.0% w/w); and level of total degradation products by HPLC (≤4.0% w/w).
Telavancin concentration (assay) is expressed as % label claim of the theoretical concentration, while the degradant concentration is determined as % w/w, which is defined as the weight of each degradant/the weight of telavancin free base in the sample. Both are determined by comparison with external telavancin reference standards injected during the HPLC analysis sequence. The purity of telavancin is expressed as % a/a, which is determined by comparing the peak area of telavancin peak with the total peak areas of all peaks in the HPLC chromatogram.
Physical Attributes
The color and clarity of the IV infusion solutions were determined by visual inspection of singular samples. Determinations of pH were performed using an accumet Basic AB15 pH meterj.
HPLC Analysis
A validated HPLC stability-indicating method was used to analyze telavancin and its impurity and degradants. The HPLC instrumentationk included a C18 columnl maintained at 30°C (86°F), a vacuum degasser, an autosampler capable of maintaining temperature at 5°C, a solvent delivery system, a column oven compartment, a diode-array detector, and the Empower softwarem. The mobile phase solutions consisted of acetonitrile:water:formic acid (2:98:0.05 v/v/v) as mobile phase A and acetonitrile:water:formic acid (60:40:0.05 v/v/v) as mobile phase B at a flow rate of 1 mL/min. The mobile phase A of the HPLC gradient was set as follows: 0 minutes, 90%; 20 minutes, 85%; 30 minutes, 80%; 50 minutes, 60%; 50.5 minutes, 0% followed with a 5-minute washout and equilibrating period. The injection volume of 80 µL was used for all injections of standards and samples. Quantitation was performed by integration of the peaks at a detection wavelength of 230 nm for telavancin and its degradant peaks.
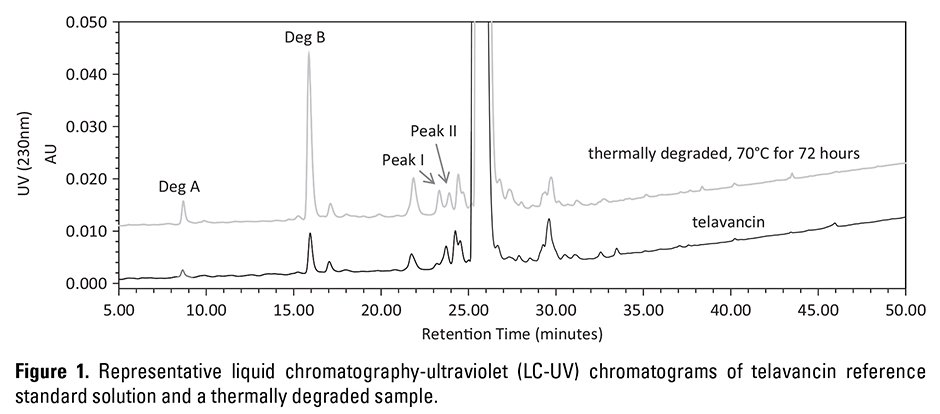
System suitability consists of a relative standard deviation (SD) of not more than (NMT) 1.0% of telavancin peak area from 6 consecutive replicate injections (80 µL) of a system suitability solution, with tailing factor of NMT 3, theoretical plates of no less than (NLT) 10,000, and resolution between peak I and peak II of NLT 0.5 (Figure 1), along with the relative SD of NMT 1% of telavancin peak area from 3 reference standard solution injections bracketing the whole injection sequence.
The HPLC method was validated in accordance with the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use.7 Method selectivity was evaluated under thermal (70°C-100°C [158°F-212°F] for 48-72 hours), solutions (base – 0.1 N sodium hydroxide, acid – 0.1 N hydrochloride, oxidative – 3% hydrogen peroxide), and photolytic (light – 1.2 x 106 lux hours) stress conditions. The impurity and degradation peaks were chromatographically separated from the telavancin peak. Figure 1 shows representative liquid chromatography-ultraviolet (LC-UV) chromatograms of a telavancin sample and a thermally degraded drug product sample stored at 70°C (158°F) for 72 hours. The retention time of the telavancin peak was approximately 25.8 minutes. The retention times of degradants A and B were at approximately 8.8 and 16.0 minutes, corresponding to relative retention time of ~0.34 and ∼0.62, respectively. Telavancin degraded substantially when exposed to acidic, basic, oxidative, photolytic, and thermal stressed conditions, but none of the degradation products interfered with the telavancin peak based on the UV peak homogeneity analysis and liquid chromatography-mass spectrometry analysis.
Linearity (r2 = 0.999) was demonstrated for a concentration range from 0.1% to 150% of the target telavancin concentration (0.2 mg/mL). The accuracy at 80% to 120% of target concentration was within 97% and 103% and the accuracy at 0.1% to 6.0% of target concentration was within 80% and 120%. The method has been shown to be specific, stability-indicating, accurate, linear, precise, reproducible, robust, and suitable for its intended use.
The excipient interference in the telavancin HPLC analysis was also examined. Telavancin placebo samples containing the same amount of hydroxypropyl-β-cyclodextrin and mannitol as the telavancin drug product were analyzed by the same method, and no overlap of these excipients with telavancin peak was observed.
RESULTS
Appearance and pH Result Summary of Solution in IV Bags
The solution appearance for all infusion solutions was clear and essentially free of particulate matter at all time points. The pH values of the drug product infusion solutions at different time points when stored at -20°C (-4°F) are shown in eTable 1. No significant (DpH < 0.6) changes in pH values over time were observed for all tested infusion solutions.
Chemical Stability of Telavancin in IV Solutions
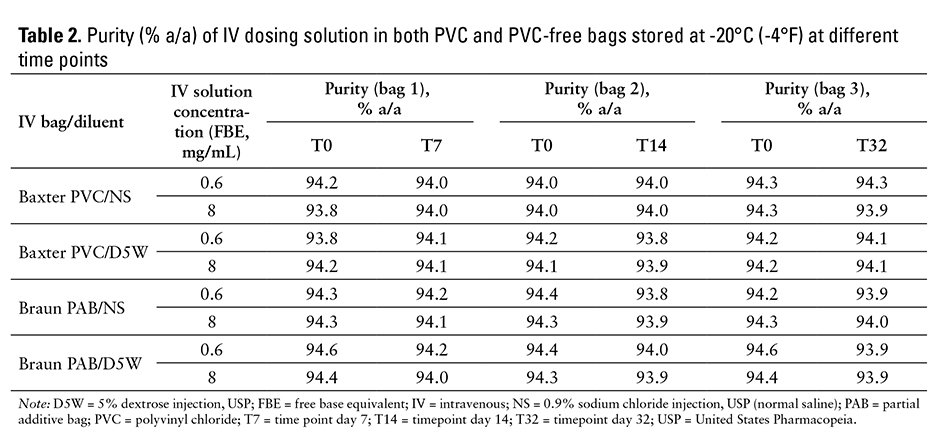
The concentrations (% label claim) of telavancin in frozen IV infusion solutions held in commercially available PVC and PVC-free IV bags at different time points are shown in eTable 2. The ratios of the concentration at various time points as compared with the initial time point are also shown in eTable 2. The concentration of each bag was compared against itself at the initial time point to minimize influence of the volume difference in the IV bags as well as to avoid any potential impact from the freeze-thaw cycle. The ratio of the telavancin concentrations at each time point tested met the predefined acceptance criteria (90.0%-110.0%), and was within 97% and 101% of the initial concentration. Due to the typical overfill (up to 15% in volume) of infusion solution bags, the actual initial concentrations of reconstituted solutions (% label claim) were up to 10% lower than the theoretical concentration (100% label claim). The purity of telavancin in frozen IV solutions at various time points is shown in Table 2, and there is no significant change in the purity of telavancin for any time point. (“Significant change” for a drug product is defined by ICH as a 5% change in concentration from its initial values or failure to meet the acceptance criteria for potency, degradation products, appearance, physical attributes, pH, and functionality test.)
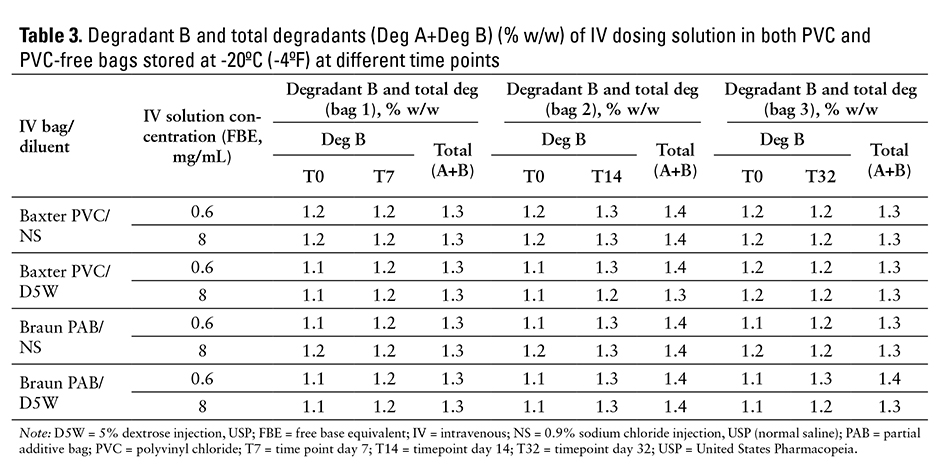
The levels of degradant A for all IV solution samples are at 0.1% w/w (as compared with acceptance criterion of NMT 1.0% w/w); data are not shown. The levels of degradant B in IV infusion solutions held in commercially available PVC and PVC-free IV bags with different IV solutions are shown in Table 3. The data on degradant B show that there is only a very slight increase in this primary degradant within 32 days when stored at -20ºC (-4ºF) conditions. The level of total degradants, which is the sum of degradant A and degradant B, for days 7, 14, and 32, is shown in Table 3; there were no significant changes. Both levels of degradant B and total degradants were well under the predefined acceptance criteria criteria (≤3% w/w for degradant B and ≤4% w/w for total degradants) during the 32 days of the stability study.
DISCUSSION
The physical and chemical stability of telavancin at therapeutic meaningful concentrations in reconstituted IV infusion solutions in PVC and PVC-free bags under frozen conditions up to 32 days were evaluated in this study. The IV infusion solutions tested in this study are those specified in the telavancin label. These data establish a reference for proper storage and handling of this drug product upon reconstitution and dilution into both PVC and PVC-free IV bags. The frozen IV bags should be brought to room temperature without any assistance, such as sonication or a hot water bath, and used immediately afterwards (within 12 hours at room temperature). The thawed solution should not be refrozen for any future use.
As with all sterile products for injection, particularly since no preservative is present in the telavancin for injection drug product, aseptic technique must be strictly observed when preparing the product for administration.
CONCLUSION
The chemical and physical stability data of telavancin IV infusion solutions in frozen condition reported here support storage at up to 32 days under frozen (-20ºC [-4ºF] or lower) conditions in either PVC or PVC-free IV bags when prepared with the diluents (either D5W or NS) tested in this study.
ACKNOWLEDGMENTS
Conflicts of interest: Dr. Gu is an employee of Theravance Biopharma US, Inc.; all other authors were employees of Theravance, Inc., the predecessor of Theravance Biopharma US, Inc., at the time of the study.
Additional contributions: The research was conducted by Theravance, Inc., the predecessor of Theravance Biopharma US, Inc. Editorial assistance was provided by Emily Hutchinson, a medical writer formerly with Envision Scientific Solutions, funded by Theravance Biopharma Antibiotics, Inc.
aTelavancin for injection, 750 mg/vial (Lot 2544-90-2029223), Ben Venue Laboratories, Inc., Bedford, OH
b5% dextrose injection, USP in ViaFlex (polyvinyl chloride) IV bags (100 mL), Baxter International Inc., Deerfield, IL, Lot #P278614
c5% dextrose injection, USP in PVC-free IV bags (100 mL), B. Braun, Bethlehem, PA, Lot # LD-216-1
d0.9% sodium chloride injection, USP (normal saline), standard ViaFlex IV bags (100 mL), Baxter International Inc., Deerfield, IL, Lot #P283408
e0.9% sodium chloride injection, USP (normal saline), in PVC-free IV bags (100 mL), B. Braun, Bethlehem, PA, Lot #LD-225-1
fSterile syringes (30 cc and 60 cc), Becton Dickinson, Franklin Lakes, NJ
gBurdick & Jackson, Muskegon, MI
hEMD, Gibbstown, NJ
iMilliQ Advantage A10, EMD Millipore, Billerica, MA
jFisher Scientific accumet Basic AB15 pH meter, Thermo Fisher Scientific, Inc. Waltham, MA
kChromatographic systems: Agilent 1100 series HPLC, Agilent Technologies, Santa Clara, CA; Waters 2695/2996 HPLC, Waters Corporation, Milford, MA
lSunFire HPLC column 3.5µm, C18, 150 x 4.6 mm, Waters Corporation, Milford, MA
mEmpower Chromatography Data System, Waters Corporation, Milford, MA
REFERENCES
- Jansen WT, Verel A, Verhoef J, Milatovic D. In vitro activity of telavancin against gram-positive clinical isolates recently obtained in Europe. Antimicrob Agents Chemother.2007;51:3420-3424.
- Krause KM, Renelli M, Difuntorum S, et al. In vitro activity of telavancin against resistant gram-positive bacteria. Antimicrob Agents Chemother. 2008;52:2647-2652.
- VIBATIV [package insert]. Theravance Biopharma Antibiotics, Inc.; 2014. http://www.vibativ.com/. Accessed August 7, 2014.
- Health Canada. Summary basis of decision (SBD) for PRVIBATIV; 2010. http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_ smd_2010_vibativ_107792-eng.php. Accessed August 6, 2014.
- Clinigen Healthcare Ltd. VIBATIV: Summary of product characteristics; 2014. http://www.ema.europa.eu/docs/en_GB/document_library/ EPAR_-_Product_Information/human/001240/WC500115364.pdf. Accessed August 6, 2014.
- Rubinstein E, Lalani T Corey GR, et al. Telavancin versus vancomycin for hospital-acquired pneumonia due to Gram-positive pathogens. Clin Infect Dis. 2011;52:31-40.
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonized Tripartite Guideline: Validation of analytical procedures: Text and methodology Q2 (R1). http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html. Accessed September 5, 2014.

Supplementary material: The online version of this article (doi: 110.1310/hpj5007-609) contains the eAppendix, including eTables 1 and 2.
*Senior Director, †Research Scientist, ‡Senior Research Associate, §Manager (II) Quality Control, Analytical Development, Technical Operations, Theravance Biopharma US, Inc., South San Francisco, California. Corresponding author: Zhengtian Gu, PhD, Senior Director, Analytical Development, Technical Operations, Theravance Biopharma US, Inc., South San Francisco, CA 94080; phone: 650-808-3746; e-mail: zgu@theravance.com