
Emerging Treatments for Heterozygous and Homozygous Familial Hypercholesterolemia
Seth J. Baum, MD, FACC, FACPM, FAHA, FNLA, FASPC,1 Daniel Soffer, MD, FNLA, FACP,2 P. Barton Duell, MD, FAHA, FNLA3
1Preventive Cardiology, Inc., Boca Raton, FL; 2University of Pennsylvania Health System, Philadelphia, PA; 3Oregon Health and Science University, Portland, OR
Familial hypercholesterolemia (FH) is an autosomal co-dominant disorder marked by extremely high low-density lipoprotein (LDL) cholesterol levels and concomitant premature vascular disease. FH is caused by mutations that most commonly affect three genes integrally involved in the LDL receptor’s ability to clear LDL particles from the circulation. Primary intervention efforts to lower LDL cholesterol have centered on therapies that upregulate the LDL receptor. Unfortunately, most patients are insufficiently responsive to traditional LDL-lowering medications. This article focuses primarily on the clinical management of homozygous FH.
[Rev Cardiovasc Med. 2016;17(1/2):16-27 doi: 10.3909/ricm0854]
© 2016 MedReviews®, LLC
Emerging Treatments for Heterozygous and Homozygous Familial Hypercholesterolemia
Seth J. Baum, MD, FACC, FACPM, FAHA, FNLA, FASPC,1 Daniel Soffer, MD, FNLA, FACP,2 P. Barton Duell, MD, FAHA, FNLA3
1Preventive Cardiology, Inc., Boca Raton, FL; 2University of Pennsylvania Health System, Philadelphia, PA; 3Oregon Health and Science University, Portland, OR
Familial hypercholesterolemia (FH) is an autosomal co-dominant disorder marked by extremely high low-density lipoprotein (LDL) cholesterol levels and concomitant premature vascular disease. FH is caused by mutations that most commonly affect three genes integrally involved in the LDL receptor’s ability to clear LDL particles from the circulation. Primary intervention efforts to lower LDL cholesterol have centered on therapies that upregulate the LDL receptor. Unfortunately, most patients are insufficiently responsive to traditional LDL-lowering medications. This article focuses primarily on the clinical management of homozygous FH.
[Rev Cardiovasc Med. 2016;17(1/2):16-27 doi: 10.3909/ricm0854]
© 2016 MedReviews®, LLC
Emerging Treatments for Heterozygous and Homozygous Familial Hypercholesterolemia
Seth J. Baum, MD, FACC, FACPM, FAHA, FNLA, FASPC,1 Daniel Soffer, MD, FNLA, FACP,2 P. Barton Duell, MD, FAHA, FNLA3
1Preventive Cardiology, Inc., Boca Raton, FL; 2University of Pennsylvania Health System, Philadelphia, PA; 3Oregon Health and Science University, Portland, OR
Familial hypercholesterolemia (FH) is an autosomal co-dominant disorder marked by extremely high low-density lipoprotein (LDL) cholesterol levels and concomitant premature vascular disease. FH is caused by mutations that most commonly affect three genes integrally involved in the LDL receptor’s ability to clear LDL particles from the circulation. Primary intervention efforts to lower LDL cholesterol have centered on therapies that upregulate the LDL receptor. Unfortunately, most patients are insufficiently responsive to traditional LDL-lowering medications. This article focuses primarily on the clinical management of homozygous FH.
[Rev Cardiovasc Med. 2016;17(1/2):16-27 doi: 10.3909/ricm0854]
© 2016 MedReviews®, LLC
KEY WORDS
Familial hypercholesterolemia • Low-density lipoprotein cholesterol • Lipid-lowering therapy • Mipomersen • Lomitapide • LDL apheresis • PCSK9 inhibitors
KEY WORDS
Familial hypercholesterolemia • Low-density lipoprotein cholesterol • Lipid-lowering therapy • Mipomersen • Lomitapide • LDL apheresis • PCSK9 inhibitors

Severe LDL cholesterol elevation is the primary mediator of atherogenesis in HeFH and HoFH.
Severe LDL cholesterol elevation is the primary mediator of atherogenesis in HeFH and HoFH.
People with HeFH respond to traditional statin and cholesterol therapies (bile acid sequestrants, niacin, cholesterol absorption inhibitors), although their responses are often blunted compared with those of the general population.
People with HeFH respond to traditional statin and cholesterol therapies (bile acid sequestrants, niacin, cholesterol absorption inhibitors), although their responses are often blunted compared with those of the general population.
Although apheresis is indicated for pediatric populations, for technical reasons associated with vascular access, it is rarely performed in individuals younger than age 5 years.
Although apheresis is indicated for pediatric populations, for technical reasons associated with vascular access, it is rarely performed in individuals younger than age 5 years.
… replacement of the LDL-R gene could potentially decrease untreated LDL cholesterol levels while also making patients with HoFH more responsive to conventional therapies.
… replacement of the LDL-R gene could potentially decrease untreated LDL cholesterol levels while also making patients with HoFH more responsive to conventional therapies.
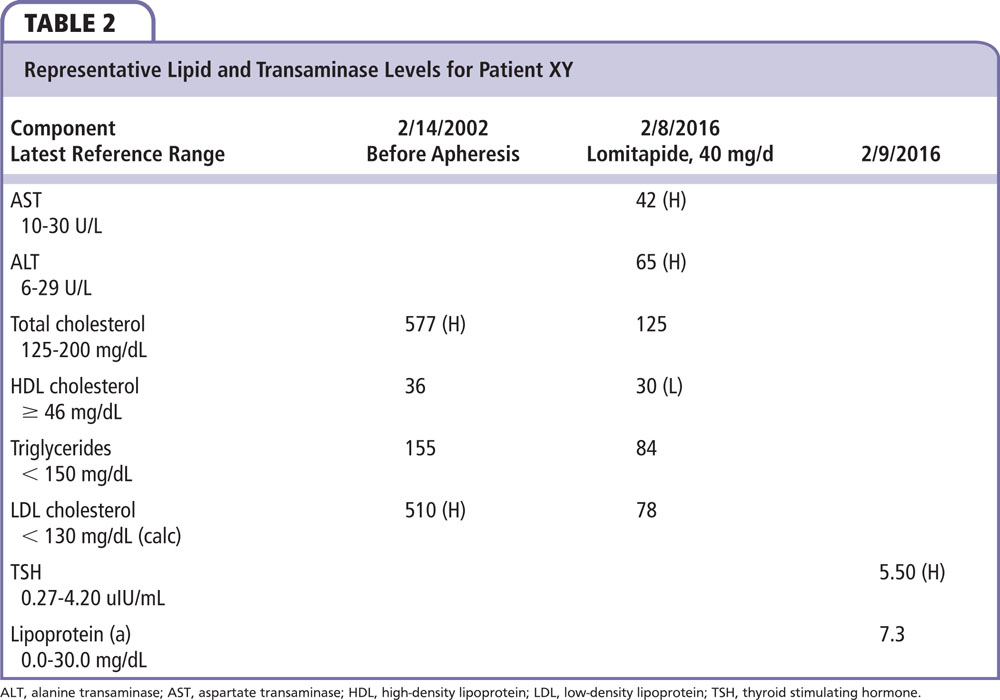
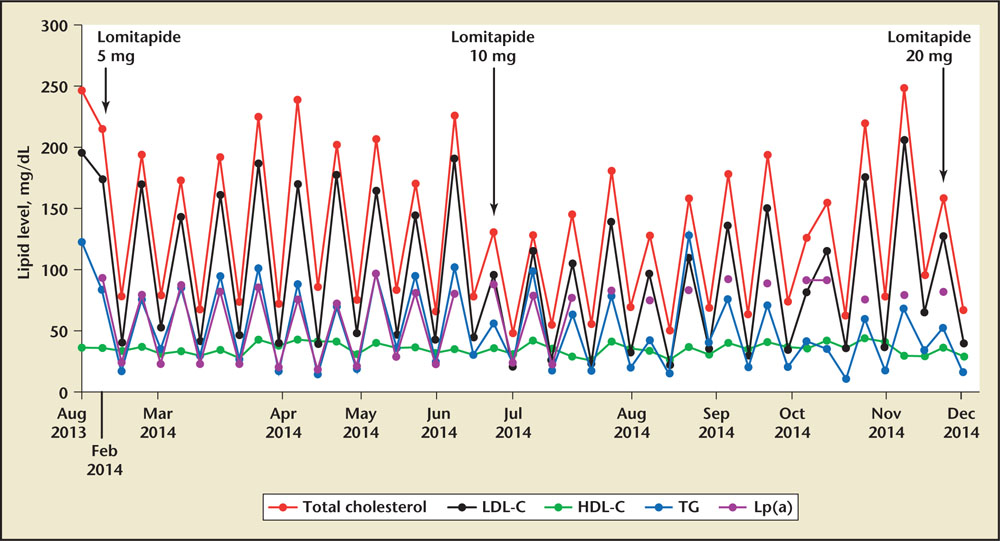
Figure 1. Lipid profiles for Patient 2 with and without lomitapide. X-axis not to scale. Peaks and troughs represent lipid levels before and after apheresis; arrows represent lomitapide dose changes according to the escalation protocol (increase in dosage) in the approved prescribing information. HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; Lp(a), lipoprotein(a); TC, total cholesterol; TG, triglycerides.
Figure 1. Lipid profiles for Patient 2 with and without lomitapide. X-axis not to scale. Peaks and troughs represent lipid levels before and after apheresis; arrows represent lomitapide dose changes according to the escalation protocol (increase in dosage) in the approved prescribing information. HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; Lp(a), lipoprotein(a); TC, total cholesterol; TG, triglycerides.
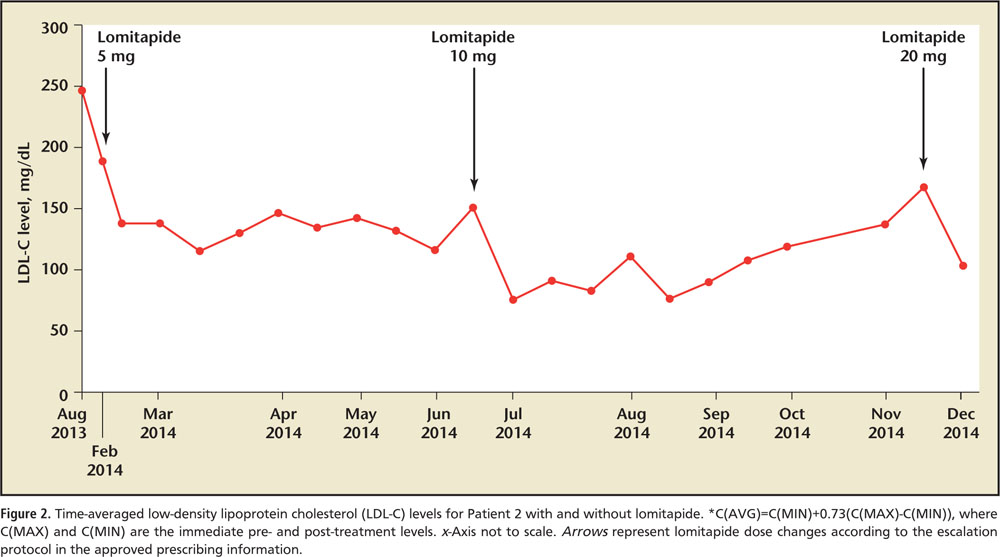
Figure 2. Time-averaged low-density lipoprotein cholesterol (LDL-C) levels for Patient 2 with and without lomitapide. *C(AVG)=C(MIN)+0.73(C(MAX)-C(MIN)), where C(MAX) and C(MIN) are the immediate pre- and post-treatment levels. x-Axis not to scale. Arrows represent lomitapide dose changes according to the escalation protocol in the approved prescribing information.
Figure 2. Time-averaged low-density lipoprotein cholesterol (LDL-C) levels for Patient 2 with and without lomitapide. *C(AVG)=C(MIN)+0.73(C(MAX)-C(MIN)), where C(MAX) and C(MIN) are the immediate pre- and post-treatment levels. x-Axis not to scale. Arrows represent lomitapide dose changes according to the escalation protocol in the approved prescribing information.
Main Points
• Familial hypercholesterolemia (FH) is a common and often undiagnosed autosomal co-dominant disorder associated with severe elevations in serum low-density lipoprotein (LDL) cholesterol and severe, premature, multivessel coronary artery disease.
• Patients should be assessed for clinical and/or genetic test options after a history and physical exam to evaluate for homozygous FH (HoFH).
• HoFH is a rare disease, occurring in approximately 1 in 160,000 individuals. Founder populations have a higher prevalence of both heterozygous FH (HeFH) and HoFH.
• Patients undergoing lipid-lowering therapy and concurrent apheresis should be evaluated to ensure appropriate LDL cholesterol levels.
• Novel therapies for HoFH (lomitapide and mipomersen) take advantage of mechanisms that do not rely on LDL receptor activity and are thus appropriate for treatment of HoFH.
• Novel therapies for HoFH (lomitapide and mipomersen) take advantage of mechanisms that do not rely on LDL receptor activity and are thus appropriate for treatment of HoFH.
• Lomitapide is an oral microsomal triglyceride transfer protein approved by the US Food and Drug Administration (FDA) for use in the management of hyperlipidemia in HoFH patients.
• Mipomersen is a subcutaneous antisense oligonucleotide injection approved for treatment of HoFH. The drug interferes with RNA translation, preventing formation of apolipoprotein B100, the main structural protein of LDL and the primary ligand for the LDL receptor.
• Alirocumab and evolocumab are proprotein convertase subtilisin/kexin type 9 inhibitors, fully human monoclonal antibody therapies administered subcutaneously every 2 or 4 weeks, approved by the FDA in 2015 as treatment for HeFH and/or atherosclerotic cardiovascular disease as an adjunct to maximally tolerated statin therapy when further LDL cholesterol reduction is required. Evolocumab has an additional indication for HoFH.
Main Points
• Familial hypercholesterolemia (FH) is a common and often undiagnosed autosomal co-dominant disorder associated with severe elevations in serum low-density lipoprotein (LDL) cholesterol and severe, premature, multivessel coronary artery disease.
• Patients should be assessed for clinical and/or genetic test options after a history and physical exam to evaluate for homozygous FH (HoFH).
• HoFH is a rare disease, occurring in approximately 1 in 160,000 individuals. Founder populations have a higher prevalence of both heterozygous FH (HeFH) and HoFH.
• Patients undergoing lipid-lowering therapy and concurrent apheresis should be evaluated to ensure appropriate LDL cholesterol levels.
• Novel therapies for HoFH (lomitapide and mipomersen) take advantage of mechanisms that do not rely on LDL receptor activity and are thus appropriate for treatment of HoFH.
• Novel therapies for HoFH (lomitapide and mipomersen) take advantage of mechanisms that do not rely on LDL receptor activity and are thus appropriate for treatment of HoFH.
• Lomitapide is an oral microsomal triglyceride transfer protein approved by the US Food and Drug Administration (FDA) for use in the management of hyperlipidemia in HoFH patients.
• Mipomersen is a subcutaneous antisense oligonucleotide injection approved for treatment of HoFH. The drug interferes with RNA translation, preventing formation of apolipoprotein B100, the main structural protein of LDL and the primary ligand for the LDL receptor.
• Alirocumab and evolocumab are proprotein convertase subtilisin/kexin type 9 inhibitors, fully human monoclonal antibody therapies administered subcutaneously every 2 or 4 weeks, approved by the FDA in 2015 as treatment for HeFH and/or atherosclerotic cardiovascular disease as an adjunct to maximally tolerated statin therapy when further LDL cholesterol reduction is required. Evolocumab has an additional indication for HoFH.
Familial hypercholesterolemia (FH) is an autosomal co-dominant disorder marked by extremely high low-density lipoprotein (LDL) cholesterol levels and concomitant premature vascular disease. Until recently, FH prevalence was believed to be 1 in 500, with an associated prevalence of homozygous FH (HoFH) of 1 in 1 million individuals, as calculated by the Hardy-Weinberg equilibrium.1 HoFH estimates remain well within typical National Institutes of Health designations of “rare” or “orphan” disorders (> 1:1500 births or < 200,000 in the United States2), but more common than classical estimates of 1 in 1 million based on the underreporting and underestimation of heterozygous FH (HeFH).
With the knowledge gleaned from a few recent studies, the prevalence of both HeFH and HoFH is much greater than previously understood; it may be as common as 1 in 200 and 1 in 160,000, respectively. Some founder populations, such as Ashkenazi Jews,3 South African Afrikaners,4 French Canadians,5 and Christian Lebanese,6 among others, have an even higher prevalence of disease.7 The founder populations represent groups who have settled in a region and have a high frequency of intermarriage (consanguinity), resulting in the perpetuation and concentration of causal mutations.
FH is caused by mutations that most commonly affect three genes integrally involved in the LDL receptor’s ability to internalize LDL particles. Known genetic defects encoding the LDL receptor (LDL-R), apolipoprotein B (apoB; familial defective apoB is now considered a subtype of FH), and proprotein convertase subtilisin/kexin subtype 9 (PCSK9) are all responsible for FH, although LDL-R defects are responsible for “classic” HoFH.8 As a result of these mutations, individuals possess LDL clearance defects that result in elevated LDL cholesterol levels.
Severe LDL cholesterol elevation is the primary mediator of atherogenesis in HeFH and HoFH. Hence, primary intervention efforts have centered on lowering LDL cholesterol. Unfortunately, most HoFH patients are insufficiently responsive to traditional LDL cholesterol–lowering medications, as they work primarily by upregulating hepatocyte LDL-R expression. LDL-Rs are absent or function suboptimally in all forms of homozygous FH, and thus their upregulation fails to elicit the same LDL cholesterol–reducing effects seen in healthy individuals. Patients with HoFH have defects in both LDL-R alleles, diminishing the capacity to upregulate LDL-R expression. The most severe HoFH patients have null mutations in both LDL-R alleles (< 2% function), and therefore can achieve only minimal lowering with traditional LDL cholesterol-lowering medications.8,9
Cascade screening has been suggested as a cost-effective diagnostic method for individuals at increased risk. The first diagnosed patient is called the index patient, or proband. His or her first-degree relatives are screened for only the detected mutation when genotyping has been performed. More commonly, clinical criteria are utilized to cascade screen relatives of the index patient. This process is repeated for the first-degree relatives (older and younger) of every newly detected person who is affected. Each child born to a parent with HeFH has a 50% chance of inheriting the disorder. For children of two HeFH parents, each child has a 25% chance of inheriting HoFH, a 50% chance of inheriting HeFH, and a 25% chance of being healthy.8,9
It is important to recognize that genotyping is insensitive, costly, and unavailable in most parts of the world. For instance, in HoFH, the diagnosis is definite if mutations are confirmed but the patient can still have HoFH if mutations are not identified. Thus, clinical history and physical examination remains the gold standard for diagnosing HoFH in many countries.
Diagnostic Criteria for FH: HeFH and HoFH
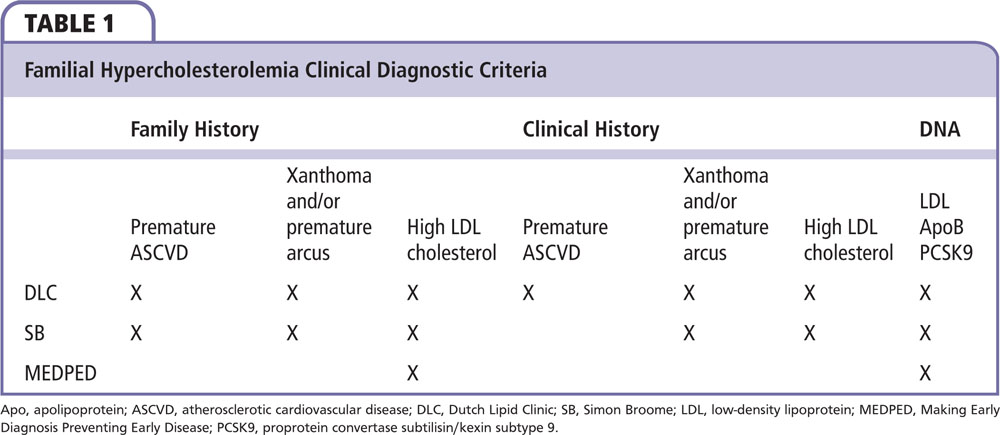
As the phenotypic expression of FH types are clearly quite broad, diagnostic criteria for HeFH and HoFH have been reconsidered. Three different criteria can help the clinician render a diagnosis of FH: (1) Dutch Lipid Clinic Network, (2) Simon Broome, and (3) MEDPED (Make Early Diagnosis Prevent Early Death).
The Dutch Lipid Clinic Network and Simon Broome criteria utilize lipid profiles, history of premature cardiovascular disease, and physical examination findings from both the patient and family. The MEDPED criteria use various total cholesterol cutoff points based on family history of FH. The Simon Broome and Dutch Lipid Clinic Network criteria consider FH to be definite, probable, or possible with presence of specific genetic mutation(s) carrying the most points (Table 1).10
The following clues on clinical evaluation point toward the diagnosis of FH:
- Premature atherosclerotic cardiovascular disease (ASCVD) and/or markedly elevated LDL cholesterol in both parents
- Less than anticipated/blunted response to lipid-lowering therapy (LLT)
- Premature ASCVD in the patient
- Tendon xanthomas
- Corneal arcus at a young age (< 45 y)
Traditional diagnostic criteria for HoFH have relied on LDL cholesterol levels in the untreated and treated patient of >500 mg/ dL and >300 mg/dL, respectively.1 However, population data have shown that approximately 30% of HoFH patients do not satisfy these criteria and there has been a marked under-diagnosis of HoFH. Additionally, it had been presumed that HoFH uniformly resulted in premature death, often a consequence of either supravalvular/valvular aortic stenosis or coronary events. We now know, however, that many patients live well past their teens, even into their 70s and beyond, especially after the advent of statins.
FH Treatments
Traditional hypercholesterolemia pharmacotherapy, which relies on the secondary effect of upregulation of the LDL-R, has either blunted or no response in HoFH (where there can be null LDL-R function), so LDL apheresis has been the foundation of treatment. People with HeFH respond to traditional statin and cholesterol therapies (bile acid sequestrants, niacin, cholesterol absorption inhibitors), although their responses may be blunted compared with those of the general population. The remainder of this article focuses primarily on the clinical management of HoFH.
LDL Apheresis
LDL apheresis, also referred to as lipoprotein apheresis, was initially approved for patients with FH and has been in use in the United States since the 1990s. It is a 2- to 3-hour procedure during which two large-bore intravenous lines are used, slowly removing and filtering a patient’s blood prior to returning it to his or her body. This is indicated as weekly therapy for LDL cholesterol lowering in individuals with ASCVD with HoFH, or every other week in individuals with HeFH (or high lipoprotein[a] levels). Present US Food and Drug Administration (FDA) approval in the United States supports its use for individuals on maximal medical therapy for at least 6 months who cannot achieve an LDL cholesterol level < 160 mg/dL if ASCVD is present, or LDL cholesterol < 300 mg/dL if no ASCVD is present. Although apheresis is indicated for pediatric populations, for technical reasons associated with vascular access, it is rarely performed in individuals younger than age 5 years.10-12 Ileal bypass, portacaval shunting, and liver transplantation can all be performed in HoFH patients; however, currently, these are not the standard of care.
Lomitapide and Mipomersen
Fortunately, newer therapies are available that work by affecting mechanisms other than LDL cholesterol upregulation and should be considered in patients with HoFH.13 The FDA approved lomitapide and mipomersen in 2012 and 2013, respectively. These medications are utilized as an adjunct to diet and other LLTs, including LDL apheresis where available. Specifically, they help reduce LDL cholesterol, total cholesterol, apoB, and non–high-density lipoprotein cholesterol in patients with HoFH; mipomersen also lowers Lp(a).14,15
Lomitapide is a small molecule oral agent that inhibits microsomal triglyceride transfer protein (MTP). MTP inhibition interrupting synthesis of hepatically derived very low-density lipoprotein (VLDL) particles (VLDL is the precursor to LDL), as well as intestinally derived chylomicrons. VLDL cholesterol and LDL cholesterol are reduced by 40% on average.16 Hepatic steatosis can occur, though its potential clinical impact remains unknown. Due to inhibition of chylomicron synthesis, steatorrhea can also occur, unless individuals adhere to very low-fat diets. Dose titration is purposefully slow in order to foster better tolerability. Lomitapide is part of the Orphan Disease Management Program; only clinicians certified by the FDA Risk Evaluation and Mitigation Strategy (REMS) program can prescribe it.
Mipomersen is an antisense oligonucleotide subcutaneous injection approved for treatment of HoFH. The drug interferes with mRNA translation, preventing formation of apoB, the main structural protein of LDL and the primary ligand for the LDL-R. Because mipomersen primarily interferes with apoB100 but not apoB48 (the truncated protein on intestinally derived chylomicrons) synthesis, it does not significantly impact intestinal chylomicron production. Mipomersen does cause hepatic steatosis at approximately the same rates as lomitapide but the levels may stabilize after the first year of treatment. Additionally, injection site reactions and flu-like reactions have limited tolerability in some patients.17 Similar to lomitapide, mipomersen can be prescribed only by clinicians certified by the REMS program. The results of a recent analysis suggest that treatment with mipomersen may reduce the incidence of cardiovascular events in patients with FH.18
Lomitapide and mipomersen have not yet been adequately evaluated in the pediatric population; therefore, these drugs do not have an FDA-approved indication for treating children.16,17 It is left to the clinician to diagnose HoFH and then weigh the risks and benefits of therapy. As noted previously, genotyping of this condition may have a false-negative result. This is related to the range of genotypes, and recognition that individuals with “classic HoFH” may be compound or double HeFH, or even HeFH with additional minor LDL cholesterol–raising gene defects. Studies have shown effectiveness of these treatments for such individuals.16,17
Gene Therapy
Gene therapy for HoFH is also being investigated as a potential treatment. This may result in improved responsiveness to conventional LDL cholesterol–lowering therapy.19
PCSK9 Inhibitors
Alirocumab and evolocumab are fully human monoclonal antibody therapies directed against PCSK9, a regulator of LDL-R activity. PCSK9 binds to the LDL-R, targeting it for lysosomal degradation instead of allowing it to recycle back to the cell membrane to promote additional LDL cholesterol uptake.20
PCSK9 inhibitors are administered as subcutaneous injections every 2 or 4 weeks. The FDA approved them in 2015 as treatment for FH and/or ASCVD, specifically as an adjunct to maximally tolerated statin therapy when further LDL cholesterol reduction is required.21 Evolocumab earned additional FDA approval for the treatment of HoFH. Both drugs have similar side-effect profiles and have been evaluated for safety in both HeFH and ASCVD patients who require additional LDL cholesterol lowering. Because of their mechanism of action, PCSK9 inhibitors appear to lack efficacy in the rare HoFH patients possessing two null receptor mutations and are moderately effective in HoFH with partial LDL-R defects.20,21
Case Reports
It is important for clinicians to recognize the spectrum of HoFH and HeFH disease. In order to qualify and receive lomitapide and mipomersen, patients must be diagnosed with HoFH. Three patient cases follow.
Case 1
Patient XY is a 30-year-old nonsmoking immigrant who was diagnosed at age 5 years with extensor surface xanthomas; her total cholesterol level was > 800 mg/ dL and she had normal triglyceride levels. Upon diagnosis, along with taking cholestyramine, her treatment included extended-release (ER) niacin, ezetimibe, and lovastatin; other statins were used over the next 20 years. She initiated LDL apheresis at age 15 years and participated in early clinical trials with lomitapide, continuing this treatment as part of the FDA compassionate use program. This treatment was converted to prescription after its FDA approval in 2012.
Despite “clean coronary arteries” at age 15, she developed angina pectoris at age 18. She then required percutaneous coronary interventions at age 20, 21, and 22 years. This was followed by three-vessel coronary artery bypass grafting with aortic valve and root replacement at 24 years, and sequential bilateral carotid endarterectomies later that same year.
This patient has tolerated lomitapide as monotherapy for more than 5 years. She has had mild transaminase elevations (<1.5× upper limit of normal) and no other side effects. Her other medications include aspirin, clopidogrel, metoprolol, and isosorbide mononitrate. Representative lipid and transaminase levels are listed in Table 2.
Mutations are present in both LDL-R alleles (delEx3-6/delEx3- 6). Her parents are second cousins. Her father died at age 62 years from complications of coronary heart disease; her mother is age 60 years and erratic with her statin adherence, but does not have clinical ASCVD. Her brother does not have high cholesterol.
The patient is recently married and has been counseled regarding her own personal safety concerns associated with pregnancy and childbirth, and needs to discontinue the lomitapide in the event of unintended pregnancy. She has an intrauterine device and she and her husband report a clear understanding of the risks of pregnancy and confirm reliable birth control.

Case 2
This patient is a 38-year-old man; he is 173 cm tall, weighs 78 kg, and has a body mass index of 26.15 kg/ m2. His physical examination reveals Achilles tendon xanthomas and a prominent corneal arcus.
The patient most likely had his first cardiac event when he was age 17. While playing basketball, he developed chest tightness and shortness of breath that subsided after 30 minutes of rest. On that occasion, no medical attention was sought. At age 23 he suffered an inferior myocardial infarction, and since then has had multiple coronary stents and a five-vessel coronary artery bypass graft. The patient’s maximum untreated LDL cholesterol level was > 400 mg/ dL and his total cholesterol has exceeded 600 mg/dL. Family history is significant for the presence of FH on both sides, as his entire parental lineage died from premature ASCVD. Both of his brothers have markedly elevated LDL cholesterol levels (one brother had a myocardial infarction at age 30) and his teenage son has an LDL cholesterol level > 300 mg/dL.
Despite combination pharmacotherapy, he had persistently elevated cholesterol levels. He therefore required the initiation of lipoprotein apheresis. As routine pre-apheresis LDL cholesterol levels have remained between 150 and 200 mg/dL, lomitapide therapy was considered. At the time lomitapide was introduced, the patient’s daily LLT regimen included rosuvastatin, 20 mg, ezetimibe, 10 mg, colesevelam, 625 mg (6 tablets), ER niacin, 500 mg, and lipoprotein apheresis every other week. The lipid response is shown in Figure 1.
The patient was started on oral lomitapide, 5 mg/d, in February 2014, then increased to 10 mg/d in July of the same year. Pre-apheresis LDL cholesterol levels then fell to approximately 130 mg/dL, and time-averaged LDL cholesterol levels fell below 100 mg/dL (Figure 1 and Figure 2). However, because of a worsening diet and exercise regimen, pre-apheresis LDL cholesterol levels slowly climbed to 248 mg/ dL in December 2014 (Figure 1). At that point the lomitapide dose was increased. Notably, he had no regression of his xanthomas. This second dose increase reduced time-averaged LDL cholesterol levels back to below < 100 mg/dL (Figure 2).

Case 3
An otherwise healthy boy developed irregular yellowish cutaneous lesions on his knees at age 5 after a normal infancy and childhood. By age 6 he started developing similar lesions on his elbows and hands. The family sought consultation with their pediatrician when their child was age 7 because new lesions were appearing and the older lesions were enlarging. A variety of tests were performed that included a LDL cholesterol measurement of 743 mg/dL. His mother’s LDL cholesterol level was 360 mg/dL and his father’s LDL cholesterol level was 250 mg/dL.
On physical examination, the patient had tuberous xanthomas involving his elbows, knees, and the intertriginous areas between his fingers. His Achilles tendons were mildly thickened. He had a I-II/VI grade systolic ejection murmur, without aortic valvulopathy or supravalvular pathology on echocardiography.
He initiated treatment with simvastatin, 20 mg/d, followed by titration up to 40 mg/d in combination with a low-cholesterol, low-saturated fat diet, which lowered his LDL cholesterol concentration from 743 mg/dL to 546 mg/ dL and 529 mg/dL, respectively. Colesevelam was added at a dose of 1.875 g twice daily, which lowered his LDL cholesterol level to 412 mg/dL. ER niacin was added and titrated up to 1 g every evening, producing an additional reduction in his LDL cholesterol to 331 mg/ dL. The addition of ezetimibe, 10 mg/d, and titration of his simvastatin dose to 60 mg/d helped lower his LDL cholesterol concentration to 308 mg/dL. The treatment regimen was subsequently changed to rosuvastatin, 40 mg/d + ezetimibe, 10 mg/d + ER niacin, 2 g/d + colesevelam, 2.5 g/d. This LLT regimen initially lowered his LDL cholesterol concentration to 178 mg/dL. However, over a 4-year period, his LDL cholesterol level progressively increased to 221 mg/ dL. By age 14, his tuberous xanthomas had resolved.
Throughout this time, the patient declined LDL apheresis. Annual stress echocardiography evaluations were initiated at age 12, which demonstrated no evidence of inducible ischemia. A coronary computed tomography (CT) angiogram was performed at age 16, which showed no areas of vascular calcification or coronary artery stenosis.
The patient had achieved a substantial reduction in his LDL cholesterol concentration from 743 mg/ dL to 221 mg/ dL in response to an aggressive four-drug regimen. However, he continued to have severe hypercholesterolemia that necessitated additional LDL lowering. Moreover, his lipoprotein(a) concentration was severely elevated to 198 mg/dL, which is associated with a doubling of risk of cardiovascular events. The addition of weekly mipomersen, 200 mg, subcutaneously lowered his LDL cholesterol to 201 mg/dL and his lipoprotein(a) to 147 mg/ dL. Lomitapide, 5 mg/d, in combination with a very low-fat diet was added to replace mipomersen. Subsequently, his lomitapide dose was titrated up to 10 mg alternating with 5 mg every other day. Although this LLT lowered his LDL cholesterol concentration to 138 mg/dL, his alanine transaminase concentration increased to 2.5 times the upper limit of normal, which necessitated a reduction in his lomitapide dose to 5 mg/d. While taking lomitapide, 5 mg/d, in combination with his other LLT, his LDL cholesterol concentration was 199 mg/dL.
The patient is currently age 23 and continues to have no evidence of myocardial ischemia on his cardiac stress echocardiogram evaluations. He recently initiated treatment with alirocumab, 150 mg, subcutaneously every 2 weeks in combination with his other five LDL-lowering medications, lowering his LDL cholesterol concentration to between 115 and 130 mg/dL.

Discussion
These case reports showcase different clinical presentations associated with HoFH. Treatment included newly approved therapies lomitapide, mipomersen, and PCSK9 inhibitors.
The patient in Case 1 has significant ASCVD and HoFH. She has known homozygous LDL-R gene mutations resulting in the classic HoFH phenotype. In addition to vigilant cardiac care with LDL cholesterol-lowering pharmacotherapy and general medical attention, she has maintained a lifelong heart-healthy diet, exercised regularly, and avoided tobacco. Of note, she had documented “clean coronary arteries” at age 15, but developed symptomatic ASCVD within a few years as is the norm for HoFH.
She receives family planning counseling at every visit because of the severity of her cardiac condition and because her medicine is Pregnancy Category X. Although her prognosis is guarded, she was offered the opportunity to participate in an LDL cholesterol gene therapy clinical trial.
The patient in Case 2 has classic early presentation with physical examination features, symptomatic ASCVD, and expected family history. Although LLT has been associated with delayed cardiovascular events and prolonged survival in patients with HoFH,1,22-25 his treatment did not adequately decrease his LDL cholesterol levels. Hence, he required LDL apheresis as adjunctive treatment.
His LDL cholesterol levels remained elevated and he was started on oral lomitapide and did not experience any adverse effects. Thus, this patient demonstrated how lomitapide can be effectively used in patients undergoing apheresis25-28 who are resistant to other LLTs.
The patient in Case 3 was treated with statins, cholesterol absorption inhibitors, bile acid sequestrants, and niacin derivatives.1,23,27 He declined apheresis despite the need for additional lowering of his cholesterol levels.
He was treated with mipomersen then switched to lomitapide because of persistent hypercholesterolemia. He was then treated with alirocumab. His LDL cholesterol decreased to a level consistent with clinical data showing LDL cholesterol reductions of 30% to 40% in HoFH patients.21,29 Thus, this patient has shown promise in the treatment of his HoFH after having less successful results from numerous other medication regimens.
Conclusions
FH (HeFH and HoFH) is a severely atherogenic disorder that can cause very early onset coronary artery disease and death. Prior to the advent of statin therapy, individuals with FH and their families faced hopelessness and despair regarding their condition. Multiple advances in treatment have occurred since the introduction of statin therapy in 1987. Medications such as lomitapide, mipomersen, and PCSK9 inhibitors have been shown to work well in patients with FH (both HeFH and HoFH [patients with severe HoFH are unresponsive to PCSK9 inhibitors]), thereby offering promise for affected individuals. Early diagnosis and aggressive treatment with medications and LDL/lipoprotein apheresis, can prevent cardiovascular complications and even allow these patients to enjoy a normal lifespan. ![]()
References
- Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. Atherosclerosis. 2012;223:262-268.
- Institute of Medicine (US) Committee on Accelerating Rare Diseases Research and Orphan Product Development. Field MJ, Boat TF, eds. Rare Diseases and Orphan Products: Accelerating Research and Development. Washington, DC: National Academies Press; 2010.
- Meiner V, Landsberger D, Berkman N, et al. A common Lithuanian mutation causing familial hypercholesterolemia in Ashkenazi Jews. Am J Hum Genet. 1991;49:443-449.
- Leitersdorf E, Van der Westhuyzen DR, Coetzee GA, Hobbs HH. Two common low density lipoprotein receptor gene mutations cause familial hypercholesterolemia in Afrikaners. J Clin Invest. 1989;84: 954-961.
- Moorjani S, Roy M, Gagné C, et al. Homozygous familial hypercholesterolemia among French Canadians in Québec Province. Arteriosclerosis. 1989;9:211-216.
- Fahed AC, Safa RM, Haddad FF, et al. Homozygous familial hypercholesterolemia in Lebanon: a genotype / phenotype correlation. Mol Genet Metab. 2011;102:181-188.
- Moorjani S, Roy M, Torres A, et al. Mutations of low density- lipoprotein-receptor gene, variation in plasma cholesterol, and expression of coronary heart disease in homozygous familial hypercholesterolaemia. Lancet. 1993;341:1303-1306.
- Marais AD. Familial hypercholesterolaemia. Clin Biochem Rev. 2004;25:49-68.
- Vogt A. The genetics of familial hypercholesterolemia and emerging therapies. Appl Clin Genet. 2015;8: 27-36.
- Hopkins PN, Toth PP, Ballantyne CM, Rader DJ; National Lipid Association Expert Panel on Familial Hypercholesterolemia. Familial hypercholesterolemias: prevalence, genetics, diagnosis and screening recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5(3 suppl):S9-S17.
- Gagné C, Gaudet D, Bruckert E; Ezetimibe Study Group. Efficacy and safety of ezetimibe coadministered with atorvastatin or simvastatin in patients with homozygous familial hypercholesterolemia. Circulation. 2002;105: 2469-2475.
- Thompson GR, Barbir M, Davies D, et al. Efficacy criteria and cholesterol targets for LDL apheresis. Atherosclerosis. 2010;208:317-321.
- Watts GF, Gidding S, Wierzbicki AS, et al. Integrated guidance on the care of familial hypercholesterolemia from the International FH Foundation. J Clin Lipidol. 2014;8:148-172.
- deGoma EM. Lomitapide for the management of homozygous familial hypercholesterolemia. Rev Cardiovasc Med. 2014;15:109-118.
- Toth PP, Shah PK, Wilkinson MJ, et al. Use of microsomal triglyceride transfer protein inhibitors in patients with homozygous familial hypercholesterolemia: translating clinical trial experience into clinical practice. Rev Cardiovasc Med. 2014;15:1-10.
- Rader DJ, Kastelein JJ. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation. 2014;129:1022-1032.
- Raal FJ, Santos RD, Blom DJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:998-1006.
- Duell PB, Santos RD, Kirwan BA, et al. Long-term mipomersen treatment is associated with a reduction in cardiovascular events in patients with familial hypercholesterolemia. J Clin Lipidol. 2016. doi: http://dx.doi.org/10.1016/j.jacl.2016.04.013. Published May 9, 2016.
- Cuchel M, Bruckert E, Ginsberg HN, et al; European Atherosclerosis Society Consensus Panel on Familial Hypercholesterolaemia. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35:2146-2157.
- Stein EA, Honarpour N, Wasserman SM, et al. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation. 2013;128: 2113-2120.
- Raal FJ, Honarpour N, Blom DJ, et al; TESLA Investigators. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385:341-350.
- Goldberg AC, Hopkins PN, Toth PP, et al; National Lipid Association Expert Panel on Familial Hypercholesterolemia. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5(3 suppl):S1-S8.
- Kolansky DM, Cuchel M, Clark BJ, et al. Longitudinal evaluation and assessment of cardiovascular disease in patients with homozygous familial hypercholesterolemia. Am J Cardiol. 2008;102:1438-1443.
- Raal FJ, Pilcher GJ, Panz VR, et al. Reduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid-lowering therapy. Circulation. 2011;124: 2202-2207.
- Nordestgaard BG, Chapman MJ, Humphries ST, et al; for the European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease. Eur Heart J. 2013;34:3478-3490.
- Cuchel M, Meagher EM, du Toit Theron H, et al; Phase 3 HoFH Lomitapide Study investigators. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381: 40-46.
- McGowan MP. Emerging low-density lipoprotein (LDL) therapies: management of severely elevated LDL cholesterol–the role of LDL-apheresis. J Clin Lipidol. 2013;7(3 suppl):S21-S26.
- Stefanutti C, Blom DJ, Averna MR, et al.; Phase 3 HoFH Lomitapide Study Investigators. The lipid-lowering effects of lomitapide are unaffected by adjunctive apheresis in patients with homozygous familial hypercholesterolaemia - a post-hoc analysis of a phase 3, single-arm, open-label trial. Atherosclerosis. 2015;240:408-414.
- Ajufo E, Rader DJ. Recent advances in the pharmacological management of hypercholesterolaemia. Lancet Diabetes Endocrinol. 2016;4:436-446.