Flecainide-induced Torsades de Pointes: Case Report and Review of Literature
Mohammad Nasser, MD, Shadi Idris, MD, Kimberly Marinelli, RN, Christian Machado, MD
Department of Medicine, Division of Cardiology, Providence Hospitals and Medical Centers, Southfield and Novi, MI
Several antiarrhythmic drugs are prone to cause QT interval prolongation and torsades de pointes (TDP). Predisposing risk factors include congenital channelopathies, severe bradycardia, drugs, and hypokalemia. Individual genetic variation and drug metabolism exaggerate susceptibility to adverse reactions. These proarrhythmic effects create a deficit in the repolarization reserve and prolong action potential duration, resulting in early afterdepolarizations, which promote a reentry circuit. Flecainide, a class IC drug, also exhibits inhibitory actions on the K+ channels, causing QT interval prolongation. We identified six cases of flecainide-induced TDP in the literature. Most patients had other predisposing factors. Bradycardia was present in all cases. Our case demonstrates two arrhythmias caused by flecainide: atrial flutter with 1:1 atrioventricular conduction and TDP. Both arrhythmias developed in the absence of hypokalemia, with the use of other drugs that prolong QT interval, or genetic predisposition. Therefore, this is purely a drug effect. This case report illustrates a rare but serious proarrhythmic property of flecainide observed particularly in women.
[Rev Cardiovasc Med. 2015;16(3):214-220 doi: 10.3909/ricm0761]
© 2015 MedReviews®, LLC
Flecainide-induced Torsades de Pointes: Case Report and Review of Literature
Mohammad Nasser, MD, Shadi Idris, MD, Kimberly Marinelli, RN, Christian Machado, MD
Department of Medicine, Division of Cardiology, Providence Hospitals and Medical Centers, Southfield and Novi, MI
Several antiarrhythmic drugs are prone to cause QT interval prolongation and torsades de pointes (TDP). Predisposing risk factors include congenital channelopathies, severe bradycardia, drugs, and hypokalemia. Individual genetic variation and drug metabolism exaggerate susceptibility to adverse reactions. These proarrhythmic effects create a deficit in the repolarization reserve and prolong action potential duration, resulting in early afterdepolarizations, which promote a reentry circuit. Flecainide, a class IC drug, also exhibits inhibitory actions on the K+ channels, causing QT interval prolongation. We identified six cases of flecainide-induced TDP in the literature. Most patients had other predisposing factors. Bradycardia was present in all cases. Our case demonstrates two arrhythmias caused by flecainide: atrial flutter with 1:1 atrioventricular conduction and TDP. Both arrhythmias developed in the absence of hypokalemia, with the use of other drugs that prolong QT interval, or genetic predisposition. Therefore, this is purely a drug effect. This case report illustrates a rare but serious proarrhythmic property of flecainide observed particularly in women.
[Rev Cardiovasc Med. 2015;16(3):214-220 doi: 10.3909/ricm0761]
© 2015 MedReviews®, LLC
Flecainide-induced Torsades de Pointes: Case Report and Review of Literature
Mohammad Nasser, MD, Shadi Idris, MD, Kimberly Marinelli, RN, Christian Machado, MD
Department of Medicine, Division of Cardiology, Providence Hospitals and Medical Centers, Southfield and Novi, MI
Several antiarrhythmic drugs are prone to cause QT interval prolongation and torsades de pointes (TDP). Predisposing risk factors include congenital channelopathies, severe bradycardia, drugs, and hypokalemia. Individual genetic variation and drug metabolism exaggerate susceptibility to adverse reactions. These proarrhythmic effects create a deficit in the repolarization reserve and prolong action potential duration, resulting in early afterdepolarizations, which promote a reentry circuit. Flecainide, a class IC drug, also exhibits inhibitory actions on the K+ channels, causing QT interval prolongation. We identified six cases of flecainide-induced TDP in the literature. Most patients had other predisposing factors. Bradycardia was present in all cases. Our case demonstrates two arrhythmias caused by flecainide: atrial flutter with 1:1 atrioventricular conduction and TDP. Both arrhythmias developed in the absence of hypokalemia, with the use of other drugs that prolong QT interval, or genetic predisposition. Therefore, this is purely a drug effect. This case report illustrates a rare but serious proarrhythmic property of flecainide observed particularly in women.
[Rev Cardiovasc Med. 2015;16(3):214-220 doi: 10.3909/ricm0761]
© 2015 MedReviews®, LLC
KEY WORDS
Flecainide • Torsades de pointes • QT prolongation • Early afterdepolarization
KEY WORDS
Flecainide • Torsades de pointes • QT prolongation • Early afterdepolarization
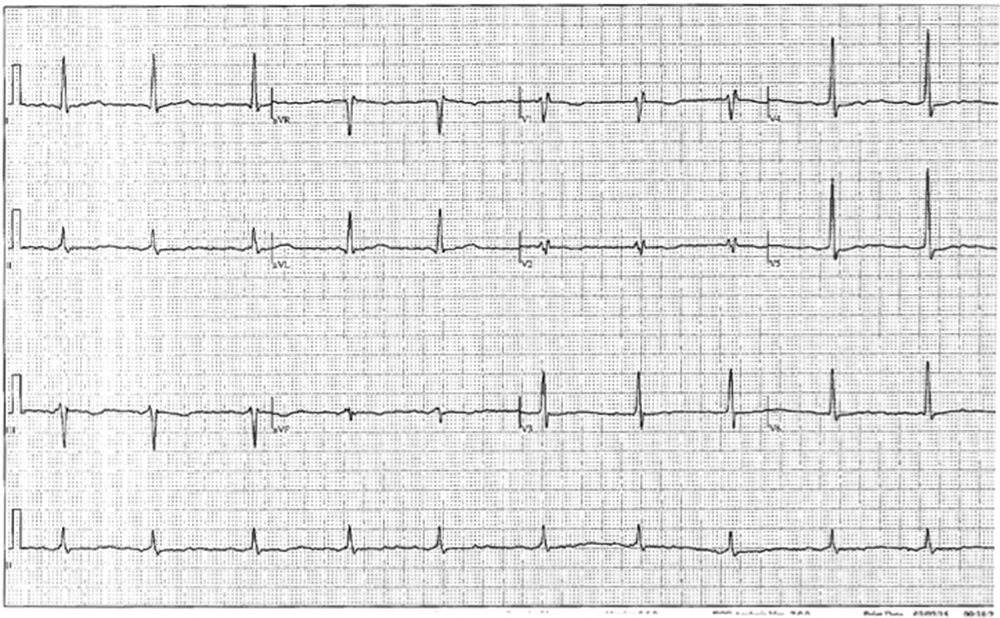
Figure 1. Pre-flecainide electrocardiogram of a 74-year-old woman with a history of paroxysmal atrial fibrillation.
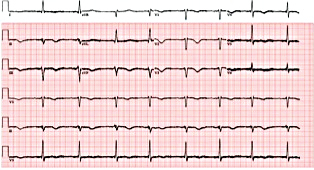
Figure 2. Electrocardiogram showing sinus bradycardia with first-degree atrioventricular block.
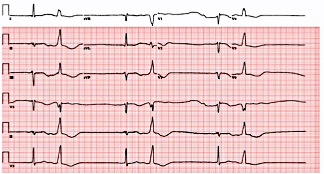
Figure 3. Heart monitor demonstrating bigeminy with prolonged compensatory pauses.
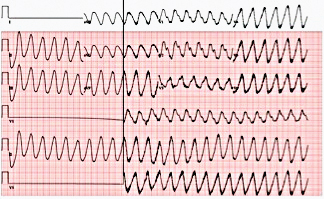
Figure 4. Electrocardiogram demonstrating atrial flutter with 1:1 atrioventricular conduction and a wide QRS complex.
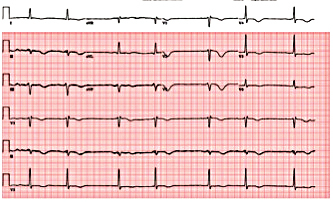
Figure 5. Electrocardiogram showing post-electrical cardioversion of atrial flutter.
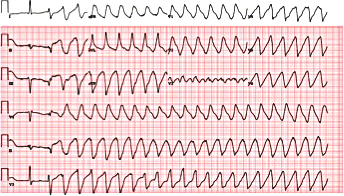
Figure 6. Electrocardiogram showing torsades de pointes.
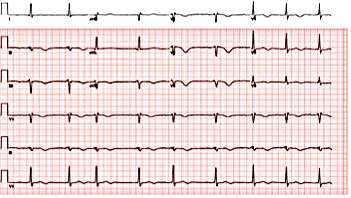
Figure 7. Electrocardiogram revealing post-electrical cardioversion of torsades de pointes.
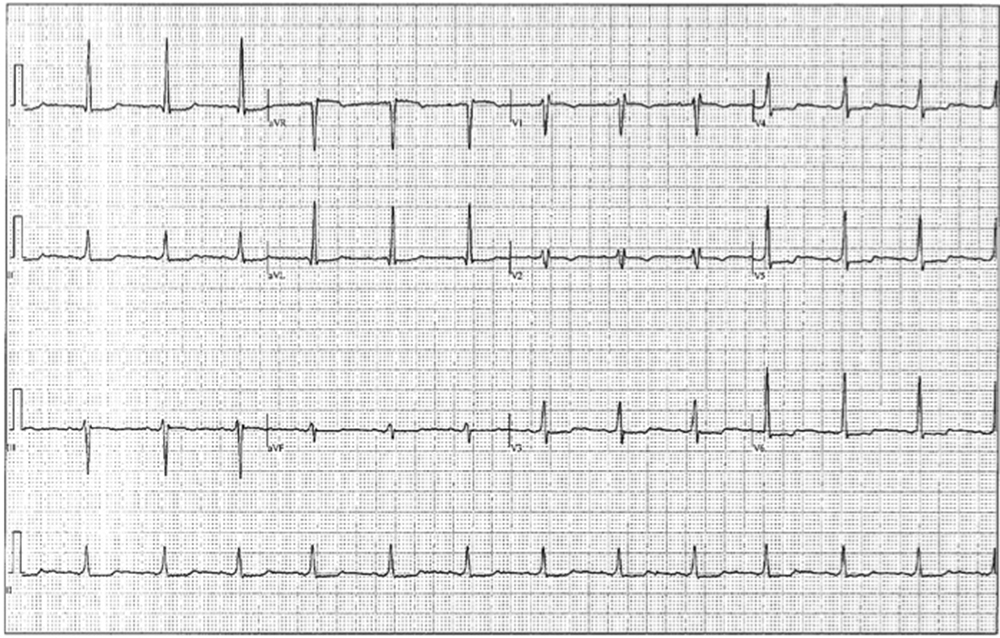
Figure 8. Post-flecainide electrocardiogram.
Studies suggest that electrophysiologic predisposition to this arrhythmia is due to early afterdepolarizations and transmural reentry mechanisms.
In recent years, more research has been conducted toward understanding the electrophysiologic properties of flecainide; in addition to its sodium channel-blocking activity, it also inhibits the L-type Ca2+ current and transient outward K channel (Ito).
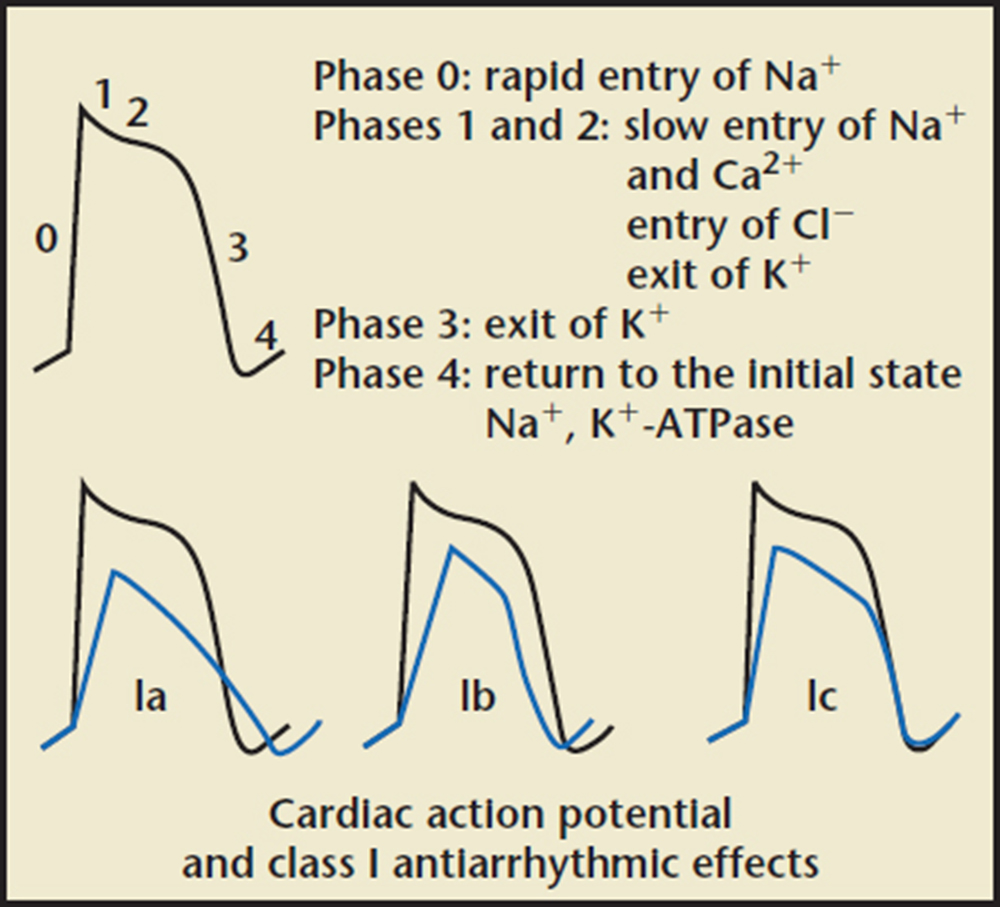
Figure 9. Action potential of class I antiarrhythmic agents. Reprinted with permission from pharmacorama.com.
Main Points
• QRS complexes with variable peaks that appear to twist around the isoelectric line characterize torsades de pointes (TDP).
• Predisposing risk factors for TDP include congenital channelopathies, severe bradycardia, drugs, and hypokalemia.
• Drug-induced TDP is of major concern because it can be life threatening; class IA/III antiarrhythmic agents, antipsychotics, antibiotics, and antihistamines are among the list of drugs that are noted to destabilize repolarization.
• Flecainide-induced TDP is rare; it is usually caused by the inhibitory effects of flecainide on the delayed-rectifier potassium channels, prolonging the QT interval, and is exacerbated by the presence of other risk factors that disturb repolarization, such hypokalemia and bradycardia.
Main Points
• QRS complexes with variable peaks that appear to twist around the isoelectric line characterize torsades de pointes (TDP).
• Predisposing risk factors for TDP include congenital channelopathies, severe bradycardia, drugs, and hypokalemia.
• Drug-induced TDP is of major concern because it can be life threatening; class IA/III antiarrhythmic agents, antipsychotics, antibiotics, and antihistamines are among the list of drugs that are noted to destabilize repolarization.
• Flecainide-induced TDP is rare; it is usually caused by the inhibitory effects of flecainide on the delayed-rectifier potassium channels, prolonging the QT interval, and is exacerbated by the presence of other risk factors that disturb repolarization, such hypokalemia and bradycardia.
Proarrhythmia or arrhythmia exacerbation is an inherent property of antiarrhythmic drugs. Class IA and class III antiarrhythmic drugs increase the action potential duration (APD) and refractoriness, leading to an expected side effect of QT prolongation, ventricular tachycardia (VT), and torsades de pointes (TDP). Class IC antiarrhythmic drugs have minimal effect on the late portion of the action potential and refractoriness, and therefore are not prominent in causing TDP. In this case report, we present a rare case of QT prolongation and polymorphic VT induced by flecainide.
Case Report
A 74-year-old woman with a history of paroxysmal atrial fibrillation (AF) was seen on a routine outpatient follow-up visit. Her medications included flecainide, 100 mg every 12 hours, and meto-prolol, 50 mg extended release daily. Before flecainide was started, her QT/QTc interval was 438/442 ms (Figure 1). During her visit, an ECG was obtained and showed AF with a rapid ventricular response of 77 beats/min. Her flecainide dose was increased to 150 mg every 12 hours. Five days later she underwent outpatient elective transesophageal echocardiography and synchronized cardioversion in an attempt to restore her sinus rhythm, which resulted in sinus bradycardia with a heart rate (HR) in the range of 50 to 60 beats/min. She had a left ventricle ejection fraction of 60%, normal systolic function, and a mildly dilated left atrium. She was continued on the same dose of flecainide and the metoprolol dose was reduced to 25 mg/d. Eleven days later, she presented to the emergency room after sustaining a syncopal episode. Upon arrival, an initial ECG revealed sinus bradycardia with a first degree atrioventricular (AV) block and a QT/QTc interval prolongation to 616/561 ms (Figure 2). Shortly thereafter, a heart monitor demonstrated frequent bigeminy complexes with prolonged compensatory pauses (1600 ms) and an HR in the range of 40 to 45 beats/min (Figure 3). She was given atropine, 0.5 mg twice, 15 minutes apart, without a significant effect. Minutes later, she became diaphoretic and unconscious, without a palpable pulse. An ECG showed atrial flutter with 1:1 AV conduction (HR 150 beats/min) (Figure 4). Electrical cardioversion was applied, converting her rhythm to sinus bradycardia (HR 42 beats/ min) with first-degree AV block and a QT/QTc interval of 640/534 ms (Figure 5). She regained consciousness without any neurologic deficits. Subsequently, her condition deteriorated once again when she lost consciousness; an ECG demonstrated sustained TDP (Figure 6). After a successful cardioversion, her rhythm was sinus bradycardia (HR 54 beats/min), first-degree AV block with a prolonged QT/QTc interval of 634/601 ms (Figure 7). Intravenous infusions of lidocaine, magnesium sulfate, and isoproterenol were started per the electro-physiologist's recommendation. Her HRincreased to > 70 beats/min and the QT/QTc interval decreased to 522/587 ms.
Additional past medical history included hypertension, gout, hypothyroidism, and osteoarthritis. She had multiple abdominal surgeries for perforated colon, cholecystectomy, and small bowel obstruction. Her medications included warfarin, metoprolol, flecainide, allopurinol, and levothyroxine. She had no history of tobacco, alcohol, or drug abuse. Her vital signs were stabilized after the arrhythmia was controlled. Physical examination did not reveal any neurologic deficits. She had a 3/6 systolic murmur best heard at the left mid sternal border. She had normal bilateral breath sounds, no lower extremity edema, and normal peripheral pulses. Laboratory testing identified the following values: leukocytes, 12.2 K/mcL; hemoglobin, 16.2 g/dL; platelets, 221 K/mcL; glucose, 213 mg/dL; creatinine, 0.9 mg/dL; sodium, 138 mmol/L; potassium, 4.3 mmol/L; magnesium, 1.5 mEq/L; calcium, 9.9 mg/dL; phosphorous, 4.2 mg/dL; alanine transaminase, 13 units/L; aspartate transaminase, 35 units/L; and thyroid-stimulating hormone, 1.47 mcIU/mL.
Flecainide was discontinued. Treatment with lidocaine and isoproterenol was continued for 24 hours. There was no recurrence of any symptoms or arrhythmia. She was discharged home in a stable condition 48 hours after admission. Two weeks later she presented to the outpatient clinic for a follow-up and an ECG was obtained, revealing a QT/QTc interval of 410/439 ms (Figure 8). She remained asymptomatic without any episodes of syncope. Genetic testing for congenital channelopathies known to cause QT prolongation was performed and the results were negative.
Discussion
Torsades de Pointes
In 1966, Francois Dessertenne referred to a polymorphic VT as TDP. QRS complexes with variable peaks that appear to twist around the isoelectric line characterize this syndrome. Associated with TDP, repolarization and QT interval abnormalities can be either acquired or congenital. The QT interval is usually prolonged beyond 500 ms. The sequence of events usually starts with premature ventricular complex, depolarizing during the termination of the QT wave, inducing short-long RR cycles precipitating TDR Predisposing risk factors include congenital channelopathies, severe bradycardia, drugs, and hypokalemia. Studies suggest that electro-physiologic predisposition to this arrhythmia is due to early afterdepolarizations (EADs) and transmural reentry mechanisms.1
Repolarization Disturbance
Cardiac myocytes are able to generate a unique action potential plateau phase, permitting Ca2+ entry into cells, a step required to replenish sarcoplasmic reticulum Ca2+ stores needed for effective contraction. During this phase, the delayed-rectifier K+ current (Ikr and Iks) is preparing to terminate depolarization. After an appropriate delay, repolarization begins with efflux of K+ out of cells. Cardiac disease and arrhythmias are well known to alter ion channel function that may initially be adaptive. For example, heart failure-induced APD prolongation by reducing the K+ current improves contractility. However, downregulation of the K+ current leads to delayed repolarization and prolongation of the plateau phase, resulting in EADs. EADs induced by derangements in delayed-rectifier currents promote formation of TDP. In addition, a decreased repolarization reserve exaggerates repolarization abnormalities by impairing the ability of myocytes to compensate for the loss of K+ current. Several studies have also linked AF to worsening delayed-rectifier K+ function and decreased mRNA expression of human ether-à-go-go-related gene (hERG), which codes for the a sub-unit of the K+ channel.2
Drug Effects
Drug-induced TDP is of major concern because it can be life threatening, with an estimated incidence of 1% to 8%. Class IA/III antiarrhythmic agents, anti-psychotics, antibiotics, and anti-histamines are among the list of drugs that are noted to destabilize repolarization. Individual genetic variation and drug metabolism exaggerate susceptibility to adverse reactions. Such drugs work at the cellular level, specifically blocking Ikr and Iks. These proarrhythmic effects create a deficit in the repolarization reserve and prolong APD, resulting in EADs, which promote a reentry circuit. This phenomenon behaves similarly to long QT syndrome. M cells and the His-Purkinje system are especially susceptible to the influence of those drugs.1
What Do We Know About Flecainide?
It is well known that class IA (quinidine, procainamide, disopyramide) and class III (amioda-rone, sotalol) antiarrhythmics are associated with a risk of developing QT prolongation. Historically, class IC drugs have been identified as weak K channel blockers, therefore weakly affecting phase 3 of the action potential (Figure 9). In recent years, more research has been conducted toward understanding the electrophysiologic properties of fiecainide; in addition to its sodium channel-blocking activity, it also inhibits the L-type Ca2+ current and transient outward K channel (Ito). Inhibitory actions on feline and guinea pig ventricular delayed-rectifier current preferentially Ikr have been described. Paul and colleagues2 examined the effects of flecainide on recombinant hERG channels eliminating contaminant currents for better accuracy. It was observed that this drug exhibits inhibitory actions on Ik with a half maximal inhibitory concentration (IC50) of 3.91 μM. Quinidine and propafenone are more potent K+ current inhibitors than flecainide.2 Inhibition of sodium channels takes place at IC50 of 1 to 2 μM. That being said, flecainide prolongs the APD in ventricular and atrial myocytes. Because flecainide has slow unbinding kinetics from Na channels, it prolongs refractoriness and slows intracardiac conduction. This effect is less pronounced in the AV node.
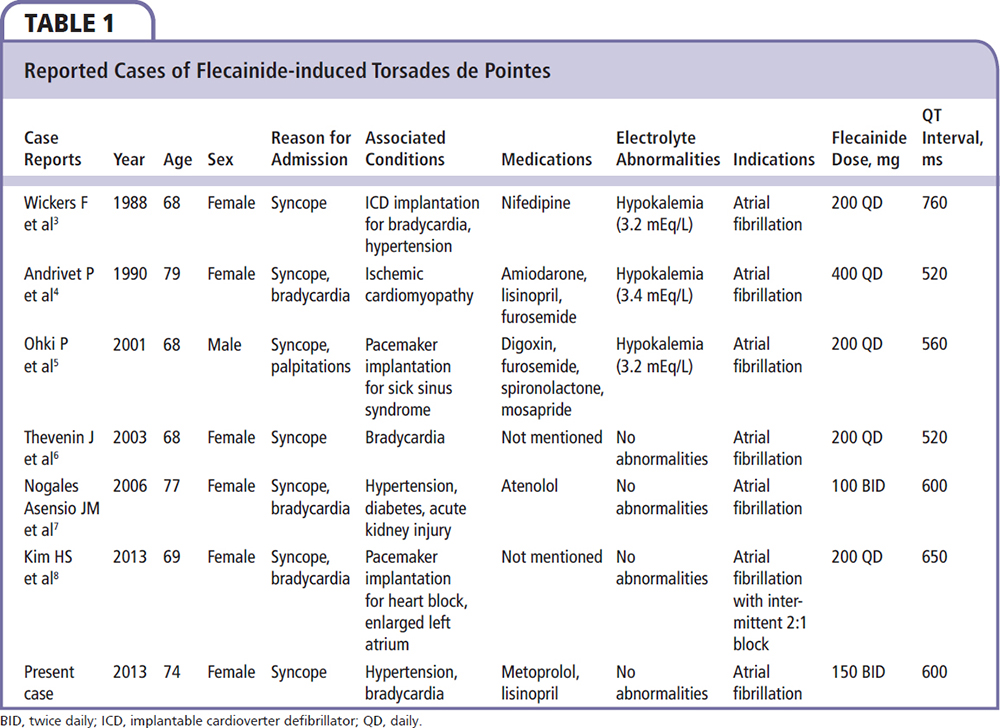
In a review of the literature, we identified six cases of flecainide-induced TDP (Table 1).3-8 Most of the patients were women. They all presented with syncope and had AF as an indication for flecainide therapy. Hypokalemia was present in half the cases, whereas bradycardia was identified in the majority of patients. Two reports described QT prolongation caused by interaction of flecainide with other drugs (amiodarone, mosapride). Our patient was a woman with paroxysmal AF. Bradycardia was present without hypokalemia. Her magnesium level was at the low end of the normal range. Test results were negative for genetic mutations known to cause long QT syndrome, indicating that this arrhythmia was a pure adverse drug effect. Therefore, it appears that women with bradycardia are especially susceptible to this arrhythmia, even in the absence of other risk factors. However, the proarrythmic effects of flecainide are more likely to be observed in synergy with other substrates that promote QT prolongation.
Our case also demonstrates a known proarrythmic effect of flecainide and class IC agents when not enough AV node blocking effect is achieved; atrial arrhythmias are slowed and potentiate a 1:1 AV conduction. Flecainide increases AV node conduction and slows down atrial rate, creating a 1:1 atrial flutter that is usually at a higher rate than 2:1 conduction. Our patient is unique in that she had two arrhythmias caused by flecainide. One is a well-described atrial flutter with 1:1 AV conduction and a wide QRS complex. Another less common arrhythmia is TDP. Both developed in the absence of hypokalemia, with the use of other drugs that prolong QT interval, or genetic predisposition.
Conclusions
Flecainide-induced TDP is a rare entity. It is usually caused by the inhibitory effects of flecainide on the delayed-rectifier potassium channels, prolonging the QT interval. This arrhythmia is exacerbated by the presence of other risk factors that disturb repolarization, such as hypokalemia and bradycardia. Presence of potassium channelopathies is not a prerequisite in this case. This report illustrates a rare but serious proarrhythmic property of flecainide. ![]()
The authors report no real or apparent conflicts of interest.
References
- Bonow RO, Mann DL, Zipes DP, Libby P. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine. 9th ed. Philadelphia, PA: Elsevier Sanders; 2012.
- Paul AA, Witchel HJ, Hancox JC. Inhibition of the current of heterologously expressed HERG potassium channels by flecainide and comparison with quinidine, propafenone and lignocaine. Br J Pharmacol. 2002;136:717-729.
- Wickers F, Haissaguere M, Palussiere J. QT prolongation and induction of torsades de pointe by flecainide. Apropos of a case [Article in French]. Arch Mal Coeur Vaiss. 1988;81:1283-1285.
- Andrivet P, Beaslay V, Canh VD. Torsades de pointe with flecainide-amiodarone therapy. Intensive Care Med. 1990;16:342-343.
- Ohki R, Takahashi M, Mizuno O, et al. Torsades de pointes ventricular tachycardia induced by mosapride and flecainide in the presence of hypokalemia. Pacing Clin Electrophysiol. 2001;24:119-121.
- Thevenin J, Da Costa A, Roche F, et al. Flecainide induced ventricular tachycardia (torsades de pointes). Pacing Clin Electrophysiol. 2003;26:1907-1908.
- Nogales Asensio JM, Moreno Sánchez N, Doncel Vecino LJ, et al. Torsade-de-pointes in a patient under flecainide treatment, an unusual case of proarrhythmicity. Int J Cardiol. 2007;114:E65-E67.
- Kim HS, Pak HN, Park JS, Kim SS. Flecainideassociated bradycardia-dependent torsade de pointes: another potential mechanism of proarrhythmia. Pacing Clin Electrophysiol. 2013;36:e84-e86.