Lomitapide for the Management of Homozygous Familial Hypercholesterolemia
Emil M. deGoma, MD
Division of Cardiovascular Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA
Homozygous familial hypercholesterolemia (HoFH) is a rare genetic disorder of low-density lipoprotein cholesterol (LDL-C) metabolism resulting in extremely elevated serum levels of LDL-C and premature atherosclerotic cardiovascular disease. Treatment typically involves multiple pharmacologic agents, as well as mechanical filtration using weekly or biweekly LDL apheresis. Despite combination lipid-lowering therapy, LDL-C levels and cardiovascular morbidity and mortality remain unacceptably high in HoFH patients. The European Commission and the US Food and Drug Administration approved the use of lomitapide, a novel medication designed to address this significant unmet need. Lomitapide is an orally administered inhibitor of microsomal triglyceride transfer protein that is indicated as an adjunct to a low-fat diet and other lipid-lowering treatments, including LDL apheresis where available for the reduction of LDL-C, total cholesterol, apolipoprotein B, and non-high-density lipoprotein cholesterol in adult patients with HoFH. The risks of transaminase elevations, hepatic steatosis, and gastrointestinal side effects, and the potential for drug interactions, require vigilant examination of the clinical and laboratory data and patient counseling prior to initiation of lomitapide, as well as regular monitoring during follow-up care. This article highlights important practical considerations for the use of lomitapide in the context of the evaluation and management of a HoFH patient case.
[Rev Cardiovasc Med. 2014;15(2):109-118 doi: 10.3909/ricm0735]
© 2014 MedReviews®, LLC
Lomitapide for the Management of Homozygous Familial Hypercholesterolemia
Emil M. deGoma, MD
Division of Cardiovascular Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA
Homozygous familial hypercholesterolemia (HoFH) is a rare genetic disorder of low-density lipoprotein cholesterol (LDL-C) metabolism resulting in extremely elevated serum levels of LDL-C and premature atherosclerotic cardiovascular disease. Treatment typically involves multiple pharmacologic agents, as well as mechanical filtration using weekly or biweekly LDL apheresis. Despite combination lipid-lowering therapy, LDL-C levels and cardiovascular morbidity and mortality remain unacceptably high in HoFH patients. The European Commission and the US Food and Drug Administration approved the use of lomitapide, a novel medication designed to address this significant unmet need. Lomitapide is an orally administered inhibitor of microsomal triglyceride transfer protein that is indicated as an adjunct to a low-fat diet and other lipid-lowering treatments, including LDL apheresis where available for the reduction of LDL-C, total cholesterol, apolipoprotein B, and non-high-density lipoprotein cholesterol in adult patients with HoFH. The risks of transaminase elevations, hepatic steatosis, and gastrointestinal side effects, and the potential for drug interactions, require vigilant examination of the clinical and laboratory data and patient counseling prior to initiation of lomitapide, as well as regular monitoring during follow-up care. This article highlights important practical considerations for the use of lomitapide in the context of the evaluation and management of a HoFH patient case.
[Rev Cardiovasc Med. 2014;15(2):109-118 doi: 10.3909/ricm0735]
© 2014 MedReviews®, LLC
Lomitapide for the Management of Homozygous Familial Hypercholesterolemia
Emil M. deGoma, MD
Division of Cardiovascular Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA
Homozygous familial hypercholesterolemia (HoFH) is a rare genetic disorder of low-density lipoprotein cholesterol (LDL-C) metabolism resulting in extremely elevated serum levels of LDL-C and premature atherosclerotic cardiovascular disease. Treatment typically involves multiple pharmacologic agents, as well as mechanical filtration using weekly or biweekly LDL apheresis. Despite combination lipid-lowering therapy, LDL-C levels and cardiovascular morbidity and mortality remain unacceptably high in HoFH patients. The European Commission and the US Food and Drug Administration approved the use of lomitapide, a novel medication designed to address this significant unmet need. Lomitapide is an orally administered inhibitor of microsomal triglyceride transfer protein that is indicated as an adjunct to a low-fat diet and other lipid-lowering treatments, including LDL apheresis where available for the reduction of LDL-C, total cholesterol, apolipoprotein B, and non-high-density lipoprotein cholesterol in adult patients with HoFH. The risks of transaminase elevations, hepatic steatosis, and gastrointestinal side effects, and the potential for drug interactions, require vigilant examination of the clinical and laboratory data and patient counseling prior to initiation of lomitapide, as well as regular monitoring during follow-up care. This article highlights important practical considerations for the use of lomitapide in the context of the evaluation and management of a HoFH patient case.
[Rev Cardiovasc Med. 2014;15(2):109-118 doi: 10.3909/ricm0735]
© 2014 MedReviews®, LLC
KEY WORDS
Atherosclerosis • Homozygous familial hypercholesterolemia • Lomitapide • Low-density lipoprotein cholesterol • Microsomal triglyceride transfer protein
KEY WORDS
Atherosclerosis • Homozygous familial hypercholesterolemia • Lomitapide • Low-density lipoprotein cholesterol • Microsomal triglyceride transfer protein
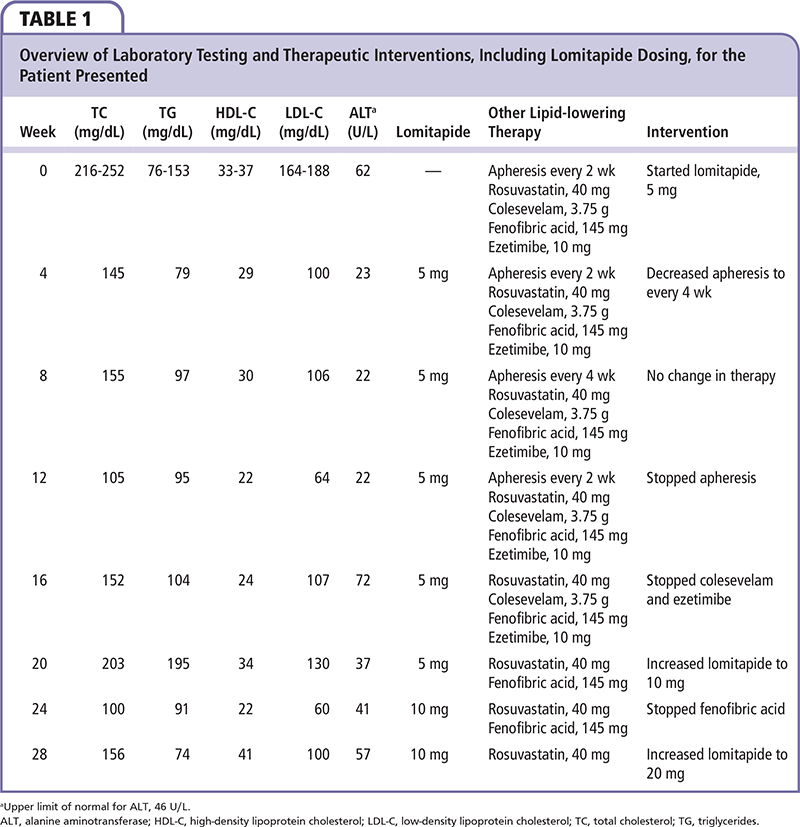
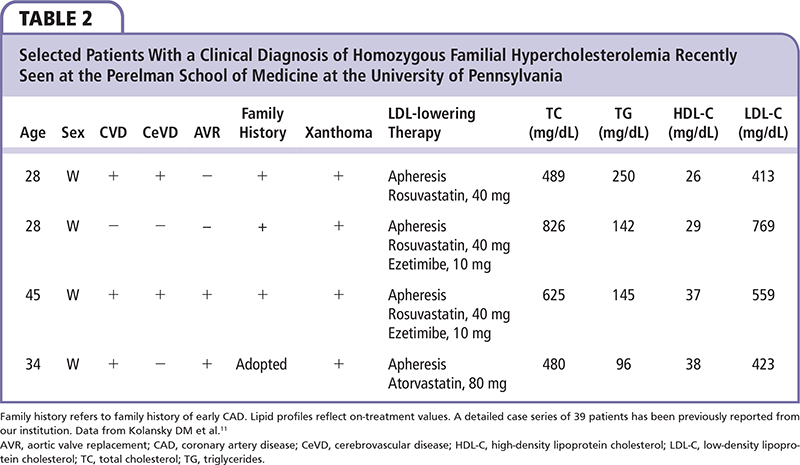
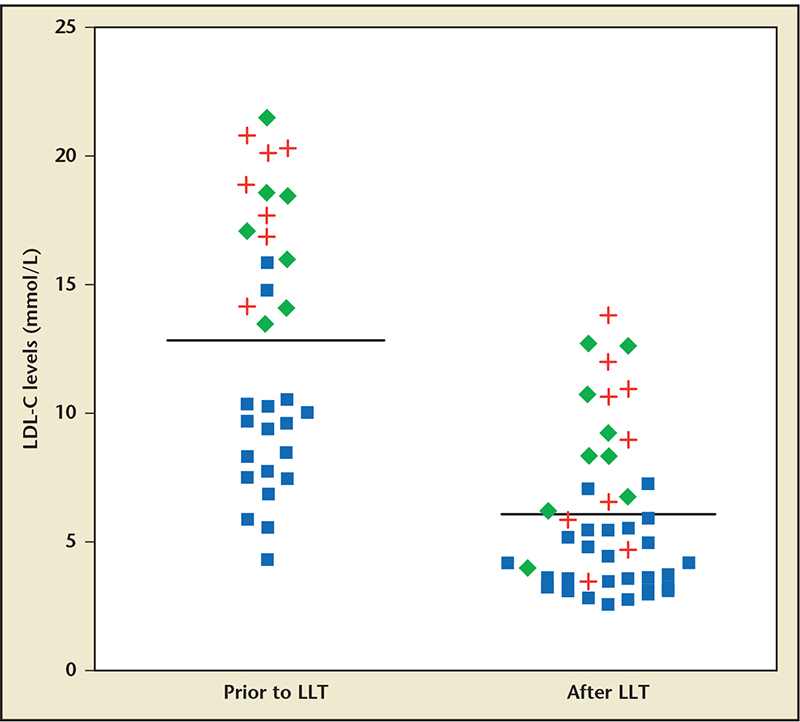
Figure 1. Low-density lipoprotein cholesterol (LDL-C) levels in patients with homozygous familial hypercholesterolemia prior to and after LDL-lowering therapy. ♦ indicates patients with two null alleles. ■ indicates patients with one null allele and one defective allele. indicates patients with two defective alleles. Horizontal lines indicate mean LDL-C levels. LLT, lipid-lowering therapy. Reproduced with permission from Sjouke B et al.4
Figure 1. Low-density lipoprotein cholesterol (LDL-C) levels in patients with homozygous familial hypercholesterolemia prior to and after LDL-lowering therapy. ♦ indicates patients with two null alleles. ■ indicates patients with one null allele and one defective allele. indicates patients with two defective alleles. Horizontal lines indicate mean LDL-C levels. LLT, lipid-lowering therapy. Reproduced with permission from Sjouke B et al.4
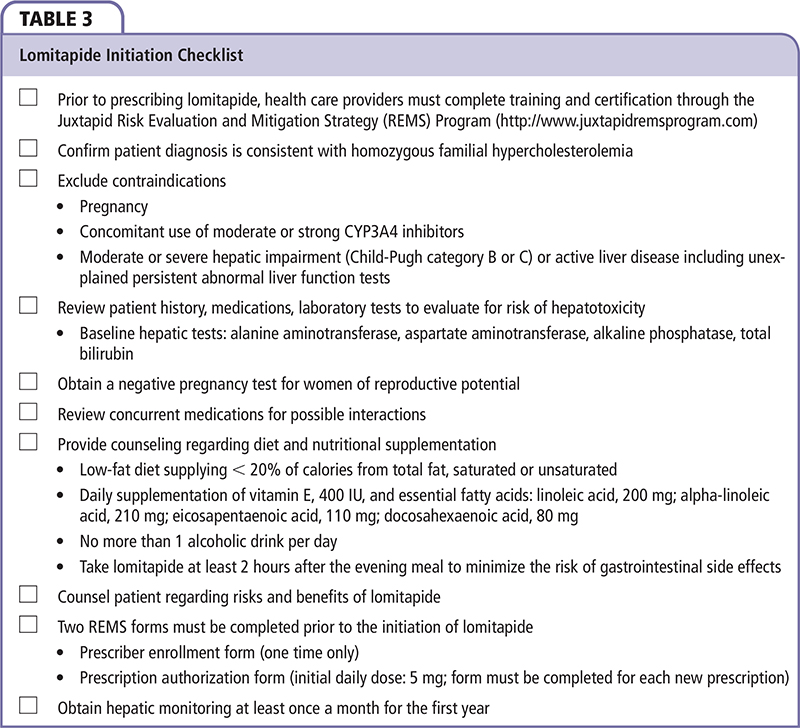
Patient counseling should describe the potential risk of hepatotoxicity; the need for regular monitoring of liver function tests (at least monthly for the first year); potential gastrointestinal side effects such as nausea, abdominal pain, and diarrhea; and dietary strategies to minimize the chance of these symptoms occurring.

Main Points
• Homozygous familial hypercholesterolemia (HoFH) is a rare genetic disorder of low-density lipoprotein cholesterol (LDL-C) metabolism. Treatment typically involves multiple pharmacologic agents, including statins and ezetimibe, and mechanical filtration using weekly or biweekly LDL apheresis.
• Despite combination lipid-lowering therapy, LDL-C levels and cardiovascular morbidity and mortality remain unacceptably high in patients with HoFH.
• Multiple clinical criteria have been published for the diagnosis of HoFH and no universal consensus has been achieved. More recent publications present individuals with genotype-confirmed HoFH who do not consistently exhibit these classic features, highlighting the phenotypic variability of HoFH.
• Lomitapide is an orally administered inhibitor of microsomal triglyceride transfer protein that is indicated as an adjunct to a low-fat diet and other lipid-lowering treatments, including LDL apheresis (where available) for the reduction of LDL-C, total cholesterol, apolipoprotein B, and non-high-density lipoprotein cholesterol in adult patients with HoFH.
• Patient counseling should describe the potential risk of hepatotoxicity; the need for regular monitoring of liver function tests (at least monthly for the first year and every 3 months thereafter); potential gastrointestinal side effects such as nausea, abdominal pain, and diarrhea; and dietary strategies to minimize the chance of these symptoms occurring.
• Withdrawal of therapy is recommended for transaminases ≥ 5 × upper limit of normal (ULN) or transaminases ≥ 3 × ULN in the setting of clinical symptoms of liver injury or concurrent elevations in bilirubin or INR. Re-challenge with lomitapide may be attempted after transaminases resolve to < 3 × ULN with consideration for dose reduction and more frequent hepatic monitoring.
• Dose reduction is recommended for transaminases ≥ 3 × ULN but < 5 × ULN in the absence of clinical symptoms of liver injury or concurrent elevations in bilirubin or INR.
• The theoretical risk of hepatotoxicity with administration warrants hepatic evaluation in the patient with new-onset gastrointestinal symptoms with liver function tests.
Main Points
• Homozygous familial hypercholesterolemia (HoFH) is a rare genetic disorder of low-density lipoprotein cholesterol (LDL-C) metabolism. Treatment typically involves multiple pharmacologic agents, including statins and ezetimibe, and mechanical filtration using weekly or biweekly LDL apheresis.
• Despite combination lipid-lowering therapy, LDL-C levels and cardiovascular morbidity and mortality remain unacceptably high in patients with HoFH.
• Multiple clinical criteria have been published for the diagnosis of HoFH and no universal consensus has been achieved. More recent publications present individuals with genotype-confirmed HoFH who do not consistently exhibit these classic features, highlighting the phenotypic variability of HoFH.
• Lomitapide is an orally administered inhibitor of microsomal triglyceride transfer protein that is indicated as an adjunct to a low-fat diet and other lipid-lowering treatments, including LDL apheresis (where available) for the reduction of LDL-C, total cholesterol, apolipoprotein B, and non-high-density lipoprotein cholesterol in adult patients with HoFH.
• Patient counseling should describe the potential risk of hepatotoxicity; the need for regular monitoring of liver function tests (at least monthly for the first year and every 3 months thereafter); potential gastrointestinal side effects such as nausea, abdominal pain, and diarrhea; and dietary strategies to minimize the chance of these symptoms occurring.
• Withdrawal of therapy is recommended for transaminases ≥ 5 × upper limit of normal (ULN) or transaminases ≥ 3 × ULN in the setting of clinical symptoms of liver injury or concurrent elevations in bilirubin or INR. Re-challenge with lomitapide may be attempted after transaminases resolve to < 3 × ULN with consideration for dose reduction and more frequent hepatic monitoring.
• Dose reduction is recommended for transaminases ≥ 3 × ULN but < 5 × ULN in the absence of clinical symptoms of liver injury or concurrent elevations in bilirubin or INR.
• The theoretical risk of hepatotoxicity with administration warrants hepatic evaluation in the patient with new-onset gastrointestinal symptoms with liver function tests.
Homozygous familial hypercholesterolemia (HoFH) is a rare genetic disorder of low-density lipoprotein cholesterol (LDL-C) metabolism resulting in extremely elevated serum levels of LDL-C and premature atherosclerotic cardiovascular disease that often manifests by the second or third decade of life.1,2 Although initial estimates reported a prevalence of 1 in 1 million,3 a recent study in the Netherlands indicated that HoFH may be at least three times more frequent, with a prevalence approaching 1 in 300,000.4 Treatment of individuals with HoFH typically involves multiple pharmacologic agents, including statins and ezetimibe, as well as mechanical filtration using weekly or biweekly LDL apheresis. Despite combination lipid-lowering therapy, LDL-C levels and cardiovascular morbidity and mortality remain unacceptably high in patients with HoFH. In December 2012 and July 2013, the US Food and Drug Administration and the European Commission, respectively, approved the use of lomitapide, a novel medication designed to address this significant unmet need. Lomitapide is an orally administered inhibitor of microsomal triglyceride transfer protein (MTP) that is indicated as an adjunct to a low-fat diet and other lipid-lowering treatments, including LDL apheresis (where available) for the reduction of LDL-C, total cholesterol (TC), apolipoprotein B, and non-high-density lipoprotein cholesterol in patients with HoFH.5-8 To highlight important practical considerations for the use of this recently approved therapy, this article discusses in detail the evaluation and management of an HoFH patient who is prescribed lomitapide.
Case Report
A 50-year-old man with premature coronary artery disease (CAD) and baseline TC levels exceeding 600 mg/dL presented for follow-up at a lipid clinic. Documentation of his untreated lipid profile was not available for review. He reported untreated triglycerides below 300 mg/dL and was taking daily lipid-modifying therapy by age 20. His first myocardial infarction occurred at age 37 and his second occurred at age 44. He underwent five percutaneous coronary interventions from age 37 to age 44, followed by three-vessel coronary artery bypass surgery at age 45. He reported stable exertional angina after walking up one to two flights of stairs. He reported no other significant medical or surgical history. His lipid-lowering therapy consisted of rosuvastatin, 40 mg/d; colesevelam, 3.75 g/d; ezetimibe, 10 mg/d; fenofibric acid, 135 mg/d; and biweekly LDL apheresis. LDL apheresis was performed via a left upper extremity fistula and recently had been associated with progressively worsening postprocedure fatigue. His other medications included aspirin, 81 mg/d; clopidogrel, 75 mg/d; carvedilol, 6.25 mg twice daily; and ranolazine, 500 mg twice daily. He had been taking ranolazine for over 1 year and had no side effects attributable to the antianginal drug. He denied the use of over-the-counter medications including acetaminophen, nonsteroidal anti-inflammatory medications, or supplements. With regard to family history, his brother was described as having TC levels exceeding 400 mg/dL with no clinical atherosclerotic disease. Lipid levels for his parents or extended family were not known. His mother died of ovarian cancer in her 40s and had no known cardiovascular disease. His father died by age 60 from cardiovascular disease. The patient denied cigarette smoking and reported consuming less than one alcoholic drink per month. Physical examination revealed bilateral Achilles tendon xanthomas and cardiovascular examination results were normal. Prior to LDL apheresis, which had been initiated 4 years ago, his LDL-C was 222 mg/dL. More recent preapheresis lipid profiles demonstrated levels of TC from 216 to 252 mg/dL, triglycerides from 76 to 153 mg/dL, high-density lipoprotein cholesterol (HDL-C) from 33 to 37 mg/dL, and calculated LDL-C from 164 to 188 mg/dL (Table 1). His liver function tests (LFTs) showed levels of serum alanine aminotransferase (ALT) at 62 U/L (upper limit of normal [ULN] 46 U/L), aspartate aminotransferase (AST) levels at 3 U/L, alkaline phosphatase at 44 U/L, total bilirubin at 0.8 mg/dL, and albumin at 3.5 g/dL. A 12-lead electrocardiogram showed sinus rhythm, no Q waves or ST-segment deviation, and a normal QT interval.
Based on the Available Clinical Information, Is the Patient a Candidate for Lomitapide?
Multiple different clinical criteria have been published for the diagnosis of HoFH, and—as yet-no universal consensus has been achieved.2-4,9-20 Published literature describes a classic presentation of HoFH to include untreated LDL-C levels > 500 mg/dL, treated LDL-C levels ≥ 300 mg/dL, tendinous and cutaneous xanthomas and corneal arcus within the first two decades of life, premature CAD and often aortic valvular disease leading to stenosis and/or regurgitation, and both a maternal and paternal history of hypercholesterolemia and atherosclerotic disease.1,2
However, more recent publications present individuals with genotype-confirmed HoFH who do not consistently exhibit these classic features, highlighting the phenotypic variability of HoFH (Table 2). In a recent study in the Netherlands, patients with genetically confirmed HoFH exhibited untreated and treated LDL-C ranging from approximately 170 to 840 mg/dL and 100 to 550 mg/dL, respectively (Figure 1).4 In a study in Quebec of 21 patients with genetically confirmed HoFH due to mutations in the LDL receptor gene LDLR, untreated TC levels varied from 491 to 1544 mg/dL.21 In a study in the United Kingdom of 15 patients homozygous or compound heterozygous for mutations in LDLR, untreated TC levels ranged from 608 to 1193 mg/dL.22 In a study in Lebanon of 18 patients with genetically confirmed HoFH due to mutations in LDLR, TC levels varied from 237 to 888 mg/dL, with an average of 547 mg/dL.23 In a recent trial of a PCSK9 inhibitor in eight patients homozygous or compound heterozygous for mutations in LDLR, pretreatment LDL-C levels ranged from 218 to 569 mg/dL.24 Similar variability in cholesterol levels as well as cardiovascular disease and the presence of xanthomas has been observed in studies of HoFH populations in South Africa,25 Italy,26 Japan,27,28 and Norway.29
A significant proportion of this variability has been attributed to the effect of the underlying LDLR mutation on LDL receptor function, whether the mutation results in deficiency of the LDL receptor (receptor-negative mutation) or a poorly functional LDL receptor (receptor-defective mutation), the latter presenting with a less severe phenotype. Among individuals with the same mutation, a large interindividual variability of LDL-C levels has been observed, pointing to additional genetic and environmental modifying factors.21,26
Although the patient's treated LDL-C levels are on the lower end of the spectrum for HoFH, his untreated cholesterol levels, tendon xanthomas, family history of hyperlipidemia (albeit with limited detail), and early and aggressive CAD support a clinical diagnosis of HoFH, acknowledging the spectrum of clinical presentations. Of note, documentation of baseline lipid levels and detailed family histories are frequently unavailable in clinical practice, as was the case for this patient, further complicating the diagnosis of HoFH.
The patient's symptoms following apheresis suggest another potential benefit in this case-reduction of the frequency or cessation of apheresis provided that adequate LDL reduction is achieved with lomitapide. In the pivotal trial of lomitapide in 29 HoFH patients, of the 13 patients on apheresis during the safety phase of the study when background therapy could be adjusted (weeks 26-78), 3 patients permanently discontinued LDL apheresis and 3 permanently increased the time interval between apheresis treatments on the basis of LDL-C response during the safety phase (weeks 26-78).6 Although the opportunity to withdraw apheresis is not guaranteed and the effect of substituting apheresis with MTP inhibition on cardiovascular outcomes remains unknown, lomitapide appears to be a particularly attractive therapeutic option to consider for HoFH patients having difficulty with regular apheresis.
What Additional Issues Should Be Addressed Prior to the Initiation of Lomitapide?
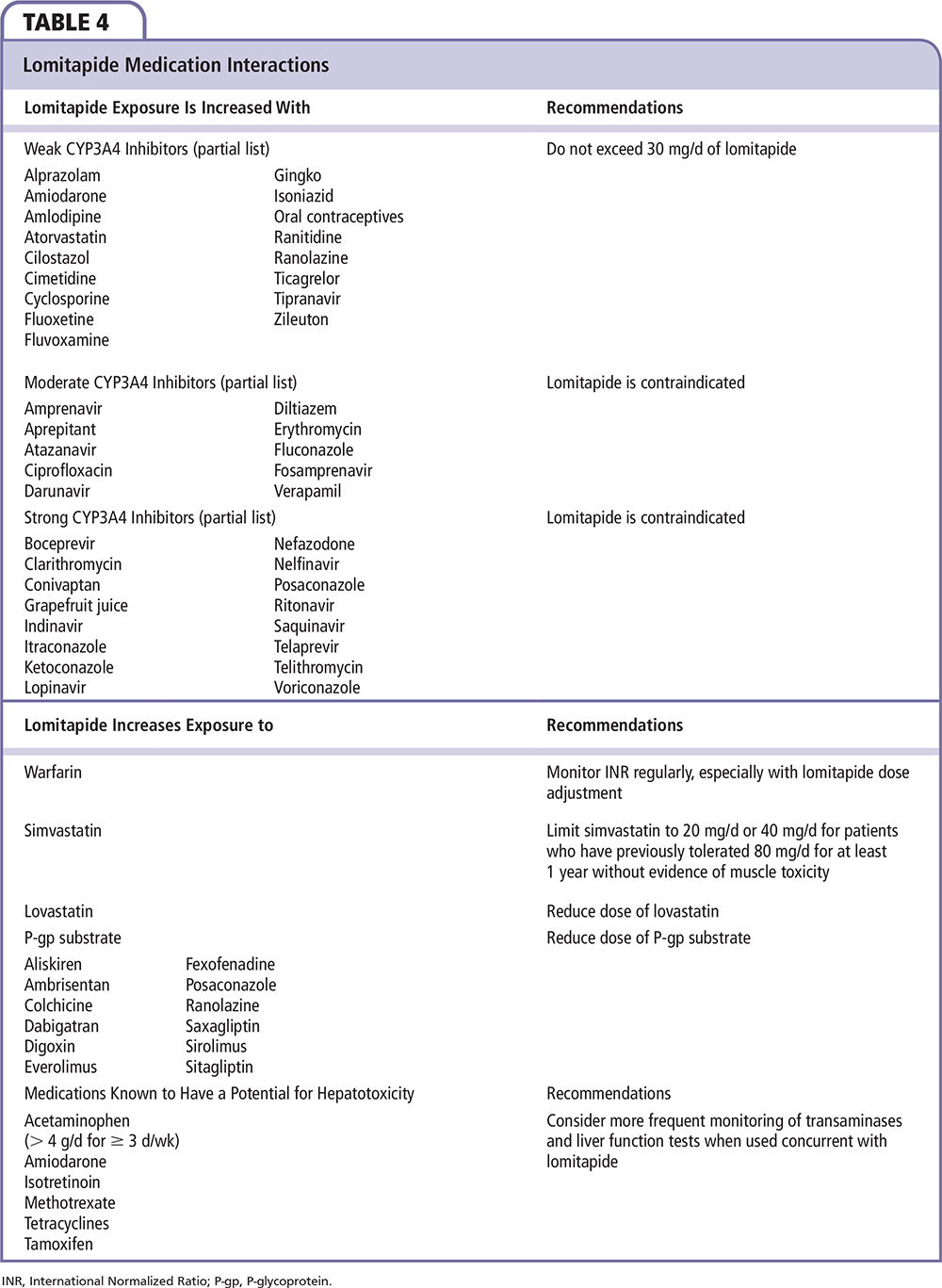
Fulfilling the criteria for a diagnosis of HoFH is only one step in determining candidacy for lomitapide and initiating the medication (Table 3). Reviewing this patient's history, medications, and laboratory tests reveals no apparent cause for a heightened risk of hepatotoxicity on lomitapide. In particular, he reported no history of liver disease, his LFTs demonstrated an isolated mild elevation of ALT < 2 × ULN, and he was not taking moderate or strong CYP3A4 inhibitors that significantly increase exposure to lomitapide. It is also important to identify any drugs whose bioavailability or pharmacodynamic effect may be enhanced by initiation of lomitapide, such as warfarin, simvastatin, lovastatin, or P-glycoprotein (P-gp) subtrates (Table 4). For the treatment of angina, he had been prescribed ranolazine, a P-gp substrate whose absorption may be increased by concomitant administration of lomitapide, a P-gp inhibitor. He reported no side effects associated with ranolazine (eg, dizziness, headaches), demonstrated a normal QT interval, and had been maintained on the starting dose of ranolazine for over a year. Taken together, this history suggests that downtitration of ranolazine was not an important consideration prior to initiating lomitapide.
Patient counseling should describe the potential risk of hepatotoxicity; the need for regular monitoring of LFTs (at least monthly for the first year); potential gastrointestinal side effects such as nausea, abdominal pain, and diarrhea; and dietary strategies to minimize the chance of these symptoms occurring (Table 3). A low-fat diet supplying < 20% of calories from total fat is recommended, along with administration of lomitapide at least 2 hours after the evening meal, in order to minimize the risk of gastrointestinal side effects. Abstinence from alcohol or minimizing alcohol use is recommended to lower the risk of steatosis. Because lomitapide may theoretically impair absorption of fat-soluble vitamins/fatty acids, supplementation is recommended to prevent nutrient deficiency.
The patient was prescribed the recommended starting dose of lomitapide—5 mg once daily—and was counseled as described. After 4 weeks, his LDL-C decreased to 100 mg/dL, and his ALT, which had previously been minimally elevated at 62 U/L, normalized to 23 U/L. At that time, the frequency of LDL apheresis was reduced from biweekly to monthly. LFT results were normal at 8 weeks. After 12 weeks, his LDL-C decreased to 64 mg/dL and apheresis was discontinued. At week 16, his LDL-C was 107 mg/dL and his ALT was elevated at 72 U/L.
What Is the Recommended Approach to ALT or AST Elevations < 3 × ULN?
In the pivotal trial for lomitapide, the incidence of at least one elevation in ALT or AST ≥ 3 × ULN was 34% and the incidence of at least one elevation in ALT or AST ≥ 5 × ULN was 14%.6 No increases in bilirubin or alkaline phosphatase were observed, and no patient discontinued lomitapide permanently because of elevations in LFT parameters.6
No additional evaluation or change in management is recommended for ALT or AST < 3 × ULN. The recommended approach to transaminasemia is detailed in Table 5. Withdrawal of therapy is recommend for transaminases ≥ 5 × ULN or transaminases ≥ 3 × ULN that do not resolve within 4 weeks or in the setting of clinical symptoms of liver injury or concurrent elevations in bilirubin or International Normalized Ratio. Rechallenge with lomitapide may be attempted after transaminases resolve to < 3 × ULN with consideration for dose reduction and more frequent hepatic monitoring.
This patient reported no gastrointestinal symptoms and most of his LFT results were normal. Because of his LDL-C response, colesevelam and ezetimibe were discontinued, and lomitapide was maintained at 5 mg/d. After 20 weeks, his LDL-C was 130 mg/dL and his ALT had normalized. Lomitapide was increased to 10 mg/d. After 3 weeks at this dose, he developed abdominal discomfort and nausea.
What Is the Recommended Approach to New-onset Gastrointestinal Symptoms?
Although no cases of symptomatic drug-related hepatitis were observed in the pivotal trial for lomitapide (n = 29),6 the theoretical risk of hepatotoxicity with administration of the MTP inhibitor warrants hepatic evaluation in the patient with new-onset gastrointestinal symptoms with LFTs. Symptomatic transaminase elevations ≥ 3 × ULN warrant withdrawal of the medication and close follow-up of LFTs (Table 5). Although the patient's symptoms should prompt immediate hepatic evaluation, abdominal discomfort and nausea more frequently occur in the absence of hepatotoxicity or other serious sequelae. The incidence of any gastrointestinal adverse reaction was 93% in the efficacy phase (first 26 weeks) of the pivotal trial of HoFH patients (diarrhea, 79%; nausea, 65%; dyspepsia, 38%; vomiting, 34%) and 73% in the safety phase (weeks 26-78). No placebo group was available for comparison. Severe gastrointestinal side effects were reported in 21% of study participants (diarrhea, 14%; vomiting, 10%; abdominal pain, 7%). Symptoms could be partially or completely controlled through adherence to a low-fat diet, administration of lomitapide timed apart from meals, and gradual dose titration.
This patient denied diarrhea or vomiting and reported adherence to a low-fat diet. LFT results were normal. No changes were made to the lomitapide dose and the symptoms resolved spontaneously after 3 days. At 24 weeks, his LDL-C declined to 60 mg/dL on rosu-vastatin, 40 mg, fenofibric acid, 145 mg, and lomitapide, 10 mg. At 28 weeks, his LDL-C and triglycer-ides were 100 mg/dL and 74 mg/dL, respectively. His ALT was 57 U/L and the remaining LFT results were normal. Fenofibric acid was discontinued and lomitapide was increased to 20 mg/d.
What Is the Recommended Approach to Lomitapide Titration and What Is the Maximum Recommended Dose for This Patient?
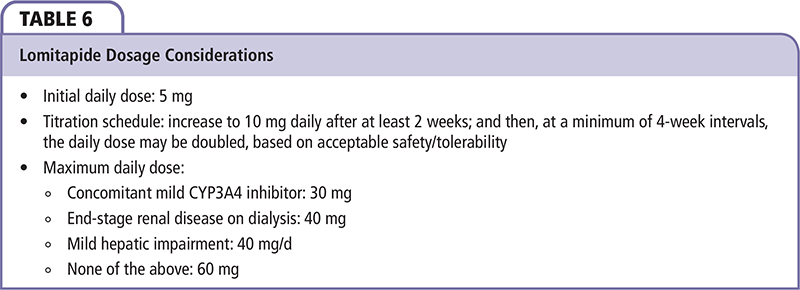
The recommended approach to lomitapide titration is summarized in Table 6. Patients should be maintained on the initial daily dose of 5 mg for at least 2 weeks, after which the dose may be doubled to 10 mg. Patients should be maintained on daily doses of 10 mg or higher for at least 4 weeks, after which the dose may be increased based on acceptable tolerability and safety. The maximum recommended dose is 30 mg in the setting of concomitant administration of a mild CYP3A4 inhibitor, 40 mg in the setting of mild hepatic impairment or end-stage renal disease on dialysis, or 60 mg absent the conditions described above. For this patient, the maximum dose is 60 mg daily; however, it is unlikely that the maximal doses will be needed to achieve acceptable LDL-C levels given his response to low-intermediate doses.
Conclusions
Lomitapide is a new addition to the therapeutic armamentarium for patients with a clinical diagnosis of HoFH. The risks of transaminase elevations, hepatic steatosis, and gastrointestinal side effects and the potential for drug interactions require vigilant examination of the clinical and laboratory data and patient counseling prior to initiation of lomitapide, as well as regular monitoring during follow- up care. ![]()
Publication of this article was made possible via sponsorship from Aegerion Pharmaceuticals, Inc. (Cambridge, MA). Editorial assistance was provided by MedReviews®, LLC. Aegerion had the opportunity to review this work for scientific accuracy and any changes resulting from comments received were made by the authors solely on the basis of scientific or editorial merit. The authors wrote and retained full control of the content of the paper.
References
- Nordestgaard BG, Chapman MJ, Humphries SE, et al; European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478-3490.
- Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. Atherosclerosis. 2012;223:262-268.
- Goldstein JL, Schrott HG, Hazzard WR, et al. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest. 1973;52:1544-1568.
- Sjouke B, Kusters DM, Kindt I, et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype-phenotype relationship, and clinical outcome [published online ahead of print February 28, 2014]. Eur Heart J. doi: 10.1093/ eurheartj/ehu058.
- Cuchel M, Bloedon LT, Szapary PO, et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med. 2007;356:148-156.
- Cuchel M, Meagher EA, du Toit Theron H, et al; Phase 3 HoFH Lomitapide Study investigators. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381:40-46.
- Cuchel M, Rader DJ. Microsomal transfer protein inhibition in humans. Curr Opin Lipidol. 2013;24:246-250.
- Juxtapid [package insert]. Cambridge, MA: Aegerion Pharmaceuticals; 2013.
- Gagné C, Gaudet D, Bruckert E; Ezetimibe Study Group. Efficacy and safety of ezetimibe coadministered with atorvastatin or simvastatin in patients with homozygous familial hypercholesterolemia. Circulation. 2002;105:2469-2475.
- Haitas B, Baker SG, Meyer TE, et al. Natural history and cardiac manifestations of homozygous familial hypercholesterolaemia. Q J Med. 1990;76:731-740.
- Kolansky DM, Cuchel M, Clark BJ, et al. Longitudinal evaluation and assessment of cardiovascular disease in patients with homozygous familial hypercholesterolemia. Am J Cardiol. 2008;102:1438-1443.
- Marais AD, Blom DJ, Firth JC. Statins in homozygous familial hypercholesterolemia. Curr Atheroscler Rep. 2002;4:19-25.
- Marais AD, Raal FJ, Stein EA, et al. A dose-titration and comparative study of rosuvastatin and atorvastatin in patients with homozygous familial hypercholesterolaemia. Atherosclerosis. 2008;197:400-406.
- Moorjani S, Roy M, Gagné C, et al. Homozygous familial hypercholesterolemia among French Canadians in Québec Province. Arteriosclerosis. 1989;9:211-216.
- Raal FJ, Pappu AS, Illingworth DR, et al. Inhibition of cholesterol synthesis by atorvastatin in homozygous familial hypercholesterolaemia. Atherosclerosis. 2000;150:421-428.
- Raal FJ, Pilcher GJ, Illingworth DR, et al. Expandeddose simvastatin is effective in homozygous familial hypercholesterolaemia. Atherosclerosis. 1997;135: 249-256.
- Raal FJ, Santos RD, Blom DJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:998-1006.
- Santos RD, Miname MH, Martinez LR, et al. Non-invasive detection of aortic and coronary atherosclerosis in homozygous familial hypercholesterolemia by 64 slice multi-detector row computed tomography angiography. Atherosclerosis. 2008;197:910-915.
- Seftel HC, Baker SG, Sandler MP, et al. A host of hypercholesterolaemic homozygotes in South Africa. Br Med J. 1980;281:633-636.
- Mabuchi H, Nohara A, Noguchi T, et al. Molecular genetic epidemiology of homozygous familial hypercholesterolemia in the Hokuriku district of Japan. Atherosclerosis. 2011;214:404-407.
- Moorjani S, Roy M, Torres A, et al. Mutations of lowdensity-lipoprotein-receptor gene, variation in plasma cholesterol, and expression of coronary heart disease in homozygous familial hypercholesterolaemia. Lancet. 1993;341:1303-1306.
- Webb JC, Sun XM, McCarthy SN, et al. Characterization of mutations in the low density lipoprotein (LDL)-receptor gene in patients with homozygous familial hypercholesterolemia, and frequency of these mutations in FH patients in the United Kingdom. J Lipid Res. 1996;37:368-381.
- Fahed AC, Safa RM, Haddad FF, et al. Homozygous familial hypercholesterolemia in Lebanon: a genotype/phenotype correlation. Mol Genet Metab. 2011;102:181-188.
- Stein EA, Honarpour N, Wasserman SM, et al. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation. 2013;128:2113- 2120.
- Raal FJ, Pilcher GJ, Panz VR, et al. Reduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid-lowering therapy. Circulation. 2011;124:2202-2207.
- Bertolini S, Pisciotta L, Rabacchi C, et al. Spectrum of mutations and phenotypic expression in patients with autosomal dominant hypercholesterolemia identified in Italy. Atherosclerosis. 2013;227:342-348.
- Yamamoto A, Harada-Shiba M, Kawaguchi A, et al. The effect of atorvastatin on serum lipids and lipoproteins in patients with homozyous familial hypercholesterolemia undergoing LDL-apheresis therapy. Atherosclerosis. 2000;153:89-98.
- Yamashita S, Ueyama Y, Funahashi T, et al. A 31-yearold woman with homozygous familial hypercholesterolemia without significant lesions in the coronary arteries. Atherosclerosis. 1986;62:117-121.
- Rodningen OK, Tonstad S, Medh JD, et al. Phenotypic consequences of a deletion of exons 2 and 3 of the LDL receptor gene. J Lipid Res. 1999;40:213-220.